Sahin Naqvi, Seungsoo Kim, Hanne Hoskens, Harold S. Matthews, Richard A. Spritz, Ophir D. Klein, Benedikt Hallgrímsson, Tomek Swigut, Peter Claes, Jonathan K. Pritchard & Joanna Wysocka
12k Accesses, 3 Citations, 34 Altmetric
Abstract
Transcriptional regulation exhibits extensive robustness, but human genetics indicates sensitivity to transcription factor (TF) dosage. Reconciling such observations requires quantitative studies of TF dosage effects at trait-relevant ranges, largely lacking so far. TFs play central roles in both normal-range and disease-associated variation in craniofacial morphology; we therefore developed an approach to precisely modulate TF levels in human facial progenitor cells and applied it to SOX9, a TF associated with craniofacial variation and disease (Pierre Robin sequence (PRS)). Most SOX9-dependent regulatory elements (REs) are buffered against small decreases in SOX9 dosage, but REs directly and primarily regulated by SOX9 show heightened sensitivity to SOX9 dosage; these RE responses partially predict gene expression responses. Sensitive REs and genes preferentially affect functional chondrogenesis and PRS-like craniofacial shape variation. We propose that such REs and genes underlie the sensitivity of specific phenotypes to TF dosage, while buffering of other genes leads to robust, nonlinear dosage-to-phenotype relationships.
Main
Transcriptional regulation is fundamental to gene expression control, and is mediated by sequence-specific TFs, a class of proteins that modulate target gene expression by binding to specific DNA motifs within noncoding REs; TFs are thus the main drivers of cellular and developmental identity1. The stability of organismal development despite environmental and genetic variation2 suggests that cellular and developmental programs are robust to modest fluctuations in TF levels. Cis-regulatory landscapes are often similarly robust, with naturally occurring genetic variation or loss of individual REs often leading to minimal effects on gene expression and/or morphology3,4,5,6.
Despite such robustness, human genetic studies have identified widespread phenotypic sensitivity to TF dosage. For instance, TFs are strongly enriched for haploinsufficient disease associations, resulting from the loss of one functional allele, and are depleted of loss-of-function variants in the general population7,8. Genome-wide association studies have revealed thousands of trait-associated variants, many of which likely act by modulating RE activity and gene expression levels9,10; trait-associated variants are also highly enriched around TF genes11,12. Both experimental and population-level data suggest that such common variants show per-allele effects on gene expression of up to 10–15% (refs. 13,14). Thus, evidence indicates that RE-driven, relatively minor variation in TF levels leads to complex trait variation, while larger dosage reductions through mechanisms such as haploinsufficiency lead to severe disorders.
Understanding how cellular and developmental programs are simultaneously robust and sensitive to TF levels is a fundamental problem, requiring quantitative studies of endogenous TF dosage effects at physiologically relevant levels. However, most studies of TF function have used knockouts, overexpression beyond trait-relevant dosage ranges, and/or genome-wide assays of unperturbed binding. Such studies have found that TFs typically regulate hundreds to thousands of REs and genes15,16,17,18, and when knocked out during development, produce pleiotropic, often embryonic lethal, phenotypes. Nonlinearity in the effects of TF dosage have been proposed to underlie TF haploinsufficiency19,20, a concept based on Fisher’s 1931 dominance model21, but such ideas have not been tested experimentally.
Transcriptional regulation is central to the development of the human face, which is key to individual identity and is disrupted in numerous craniofacial disorders that together account for approximately one-third of birth defects22. Much of both normal-range and disease-associated variation in facial shape derives from cranial neural crest cells (CNCCs), a transient, embryonic cell population that arises from the neural folds and migrates to the developing facial prominences, giving rise to most of the craniofacial skeleton and connective tissue23. Our recent review of human craniofacial genetics found that TF-encoding loci are frequently involved in both common (influencing normal-range shape) and rare (causative for Mendelian, haploinsufficient disorders) variation24. Thus, studying the quantitative effects of TF dosage alterations in craniofacial development could provide general insights into mechanisms underlying dosage sensitivity and/or robustness.
Multiple lines of evidence highlight the developmentally important TF SOX9 as an attractive model for studying TF dosage. Heterozygous loss-of-function mutations in SOX9 cause campomelic dysplasia, a disorder manifesting in long bone and sex determination defects, and a set of craniofacial features termed Pierre Robin sequence (PRS), characterized by underdevelopment of the lower jaw (micrognathia)25,26. These observations suggest that among the diverse cell types regulated by SOX9 (reviewed in ref. 27), CNCCs, chondrocytes and Sertoli cells exhibit heightened sensitivity to about 50% SOX9 dosage reduction. PRS without long bone defects can be caused by heterozygous deletion of CNCC-specific enhancers of SOX9 (refs. 28,29), whereas common genetic variants in noncoding regions near SOX9 are associated with normal-range facial variation in individuals of primarily European and East Asian ancestry30,31,32. Furthermore, CNCC-specific perturbations in mice revealed that craniofacial development is sensitive to Sox9 dosage changes over a broad range29, with even 10–13% reduction in Sox9 mRNA levels producing a subtle but reproducible change in lower jaw morphology29.
Here we sought to understand the response to quantitative changes in SOX9 dosage at multiple levels: chromatin, gene expression, cellular phenotypes and facial morphology. We applied the degradation tag (dTAG) system to achieve tunable modulation of SOX9 dosage in an in vitro model of human CNCC development. We found RE chromatin accessibility to be broadly buffered against small to moderate changes in SOX9 dosage, with a subset of REs associated with specific regulatory features showing heightened sensitivity. Gene expression shows a similar, primarily buffered, response to SOX9 dosage, with a subset of sensitive genes; these responses can be partially predicted from chromatin accessibility. Pro-chondrogenic genes, in vitro chondrogenesis itself, and genes and REs associated with PRS-like phenotypes exhibit heightened sensitivity to SOX9 dosage. We propose a model in which dosage-sensitive REs and genes transmit quantitative TF dosage changes to specific cellular and morphological phenotypes, while other phenotypically important REs and genes are regulated by SOX9 but highly buffered and are thus robust to dosage.
Results
Precise modulation of SOX9 dosage in hESC-derived CNCCs
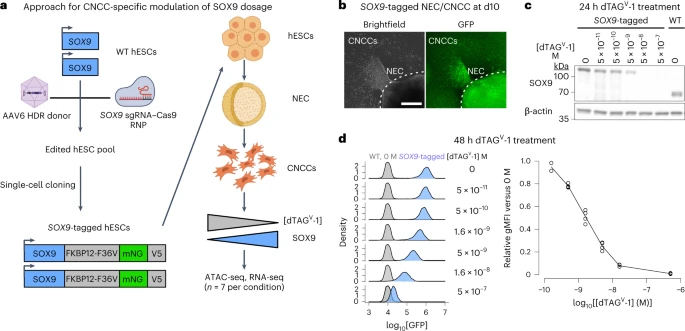
On the basis of reports that the dTAG system could be used for rapid or tunable target degradation33,34,35, we sought to apply dTAG to modulate SOX9 dosage in human embryonic stem cell (hESC)-derived CNCCs. Our approach involves genome editing in hESCs to tag SOX9 with FKBP12-F36V, which mediates degradation following addition of a heterobifunctional molecule (dTAGV-1), the fluorescent protein mNeonGreen as a quantitative proxy for SOX9 levels, and the V5 epitope for biochemical assays. Using a selection-free genome editing method36, we obtained two hESC clones with biallelic knock-in of the FKBP12-F36V–mNeonGreen–V5 tag at the SOX9 carboxy terminus (Extended Data Fig. 1a).
To avoid indirect effects of depleting SOX9 during hESC-to-CNCC differentiation, we first differentiated SOX9-tagged hESCs using an established protocol that yields molecularly nearly homogenous CNCCs37,38, and subsequently titrated SOX9 levels by adding different dTAGV-1 concentrations (Fig. 1a). Differentiation of SOX9-tagged hESCs revealed nuclear fluorescence in a subset of cells within neuroepithelial spheres and in early-stage migratory CNCCs (Fig. 1b), consistent with known roles of SOX9 in CNCC specification and migration39,40. Later-stage SOX9-tagged CNCCs showed similar SOX9 levels as untagged (wild type (WT)) CNCCs (Fig. 1c), and absolute SOX9 levels between the two SOX9-tagged clones were very similar (Extended Data Fig. 1b). Treating SOX9-tagged CNCCs with a tenfold dilution series of dTAGV-1 for 24 h yielded a gradual change in SOX9 levels (Fig. 1c). Optimization of dTAGV-1 concentrations and 48-h treatment yielded six distinct and reproducible SOX9 concentrations (Fig. 1d, right). Single-cell fluorescence quantification revealed a unimodal distribution that shifted to lower signals with higher dTAGV-1 concentrations, indicating uniform effects of dTAGV-1 despite some heterogeneity in SOX9 levels (Fig. 1d, left). Together, these results indicate that dTAG can be used to precisely modulate SOX9 dosage in hESC-derived CNCCs.
Discussion
Here we have quantified the relationship between TF dosage and phenotype at molecular, cellular and morphological levels, using SOX9 as a model. To synthesize our observations, we propose a model (Fig. 7a) in which REs regulated by SOX9 range from sensitive to buffered as a result of their cis-encoded features that determine the mode and level of binding by SOX9 and other key CNCC TFs. Genes with nearby sensitive REs will themselves show more sensitive responses to SOX9 dosage, while those with nearby buffered REs are more robust. Genes with generally important roles in CNCC biology but causing phenotypes distinct from those associated with SOX9 are buffered against SOX9 dosage change, but a subset of sensitive genes impacts specific cellular processes and morphological features similar to those associated with SOX9. Specifically, we find that several key pro-chondrogenic genes and in vitro chondrogenesis are sensitized to SOX9 dosage. Thus, the observed sensitivity of both chondrogenic effector genes and chondrogenesis itself could account for the specificity of SOX9-associated, PRS-like mandibular phenotypes, perhaps via effects on Meckel’s cartilage, a cartilage ‘template’ involved in mandible formation51.