Shohei Kojima, Satoshi Koyama, Mirei Ka, Yuka Saito, Erica H. Parrish, Mikiko Endo, Sadaaki Takata, Misaki Mizukoshi, Keiko Hikino, Atsushi Takeda, Asami F. Gelinas, Steven M. Heaton, Rie Koide, Anselmo J. Kamada, Michiya Noguchi, Michiaki Hamada, Biobank Japan Project Consortium, Yoichiro Kamatani, Yasuhiro Murakawa, Kazuyoshi Ishigaki, Yukio Nakamura, Kaoru Ito, Chikashi Terao, Yukihide Momozawa & Nicholas F. Parrish
8723 Accesses, 63 Altmetric
Abstract
Mobile genetic elements (MEs) are heritable mutagens that recursively generate structural variants (SVs). ME variants (MEVs) are difficult to genotype and integrate in statistical genetics, obscuring their impact on genome diversification and traits. We developed a tool that accurately genotypes MEVs using short-read whole-genome sequencing (WGS) and applied it to global human populations. We find unexpected population-specific MEV differences, including an Alu insertion distribution distinguishing Japanese from other populations. Integrating MEVs with expression quantitative trait loci (eQTL) maps shows that MEV classes regulate tissue-specific gene expression by shared mechanisms, including creating or attenuating enhancers and recruiting post-transcriptional regulators, supporting class-wide interpretability. MEVs more often associate with gene expression changes than SNVs, thus plausibly impacting traits. Performing genome-wide association study (GWAS) with MEVs pinpoints potential causes of disease risk, including a LINE-1 insertion associated with keloid and fasciitis. This work implicates MEVs as drivers of human divergence and disease risk.
Main
MEs characteristically insert copies of themselves into new genome locations. The evolutionary innovations of MEs are constrained within the linear descent of their host genomes; thus, differences in the sequences, mobilization activity or insertion preferences of the MEs in a particular lineage can increase the rate at which descendant genomes accumulate mutations characteristic of that lineage. In other words, MEs can accelerate genomic divergence. MEs account for a large part of species-specific genomic differentiation1, but the degree to which MEs cause species-level phenotypic differences is difficult to dissect due to accumulation of other genetic variation. MEs may also be a force driving speciation, but direct evidence of within-species divergence driven by MEs is limited2.
MEs influence the complex traits that differentiate humans and human populations, but our view of this landscape remains partial. Insertions of each of the MEs actively replicating in human genomes—namely, long interspersed nuclear element 1 (L1), SINE-VNTR-Alu (SVA) and Alu elements—have been implicated in Mendelian diseases3. For example, an SVA insertion so far only reported in the Japanese population causes Fukuyama congenital muscular dystrophy4. Individuals carrying a SLCO1B3 allele with exonic insertion of a proposed Japanese-specific highly active L1 (ref. 5) develop a benign form of hyperbilirubinemia6. Recent studies have identified ME polymorphisms associated with differential gene expression7,8,9,10 and differential polygenic disease risk11,12, but the global influence on human traits remains unclear. MEs make up a large fraction of DNase hypersensitive sites13, which are enriched in complex trait heritability14, and are also the main source of novel regulatory elements in primate genomes15. Moreover, SVs, about a quarter of which are MEVs16,17, are frequently in tight linkage disequilibrium (LD) with eQTL and trait-associated variants17,18. Actively replicating MEs necessarily carry promoters and transcription-factor binding sites that drive their expression, and some MEs appear to have been coopted as lineage-specific gene regulatory elements19,20. These observations provide a rationale to comprehensively assess the impact of ME polymorphisms on gene expression and complex traits, for example, by performing ME-oriented genotype-trait association studies.
One barrier to ME-phenotype correlation is the low accuracy of current methods used to genotype MEVs, lower than those available for single-nucleotide variants (SNVs) and often too low to derive meaningful hypotheses from statistical genetics approaches. Long-read and strand-specific sequencing are ideal to resolve MEVs and other SVs17,21; however, the number of genomes studied using these methods is low and will remain orders of magnitude lower than those genotyped by short reads until new enabling technologies emerge22,23.
Results
Development and benchmarking of MEGAnE
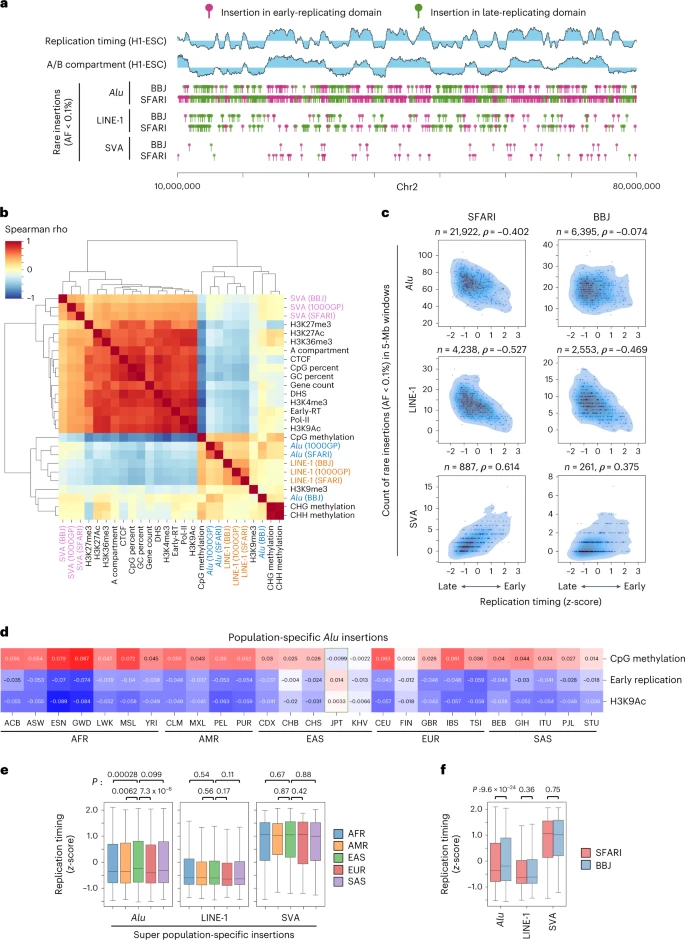
Accurate variant genotyping is required for statistical genetics. To enable both discovery and accurate MEV genotyping from genomes studied using short reads, we developed a new bioinformatic tool, mobile element genotype analysis environment (MEGAnE; Supplementary Note). Here, we interrogated the consequences of recent ME activity on human genomes and phenotypes. Accurate detection of MEVs in diverse human populations allowed us to resolve population-specific patterns of recent genome diversification accounted for by ME insertions. These may reflect different active ME copies34 or differences in the repertoire of factors repressing MEs. Although Alu insertions tend to be observed in late-replicating domains, this trend was mitigated in East Asians and even reversed in Japanese. This finding suggests that the insertion preference of Alu has shifted as humans have populated the earth. Previous work suggested a similar drift in insertion preference occurred during primate radiation; older, nonpolymorphic Alu are known to be enriched in early-replicating domains, whereas recent polymorphic ones show the opposite trend35. The factors besides ORF2p that regulate the insertion preferences of human MEs are unknown; changes to the spatiotemporal regulation of transposition-competent ribonucleoproteins could result from accumulation of population-specific mutations in these factors or in active MEs themselves.
Our ME-eQTL analyses shed light on the complex but coherent regulatory logic encoded by MEVs. Although 3’UTR Alu are often detected as multi-tissue eQTLs, some are clearly tissue-specific, such as Alu-MAP3K21 specific to the brain. Context (for example surrounding sequence and co-expressed genes) is decisive in licensing Alu polymorphisms to exert post-transcriptional regulation. Consistent with this concept, we identified FAM120A as a co-regulator of 3’UTR Alu. Disruption of interactions like that of FAM120A could represent a new target for multipurpose precision medicines. The 3’UTR Alu MEV in HSD17B12 causes changes in reporter gene expression and associates with a number of biometric traits and basal metabolic rate (highlighted in Supplementary Table 13); this variant can thus be considered to causally influence human weight, and blocking this Alu’s regulatory effect can be predicted to be tolerated. Similarly, a 3’UTR Alu in the SARS-CoV-2 host factor and dementia-linked gene TMEM106B36,37, detected as an ME-eQTL in several tissues, is associated with a number of mental health phenotypes (highlighted in Supplementary Table 13). It will be of great interest to define additional class-specific regulatory effects of MEVs, as these will advance the interpretability of non-coding genomic variation.
Inclusion of MEVs in GWAS bridges the gap between known risk loci and underlying genetic causes, demonstrating a new path to overcome the challenge of connecting GWAS signals in non-coding regions to causal variants, especially in non-European populations. By accurately genotyping MEVs and determining their linkage with SNVs, we identified hundreds of MEVs present on known risk haplotypes. These include an L1 insertion we show is causal for altered gene expression and potentially mediates the increased keloid risk associated with this haplotype. The mechanism demonstrated in the case of L1-NEDD4—that an intronic L1 insertion observed as an ME-eQTL enhances gene/isoform expression to potentially drive pathogenesis—represents an attractive hypothesis for a class of ME-trait associations we document. For example, an L1 insertion in an intron of the gene encoding thyroid stimulating hormone receptor (TSHR) and also detected as a TSHR ME-eQTL is associated with Graves’ disease (characterized by TSHR-reactive autoantibodies), and an L1 insertion intronic to and associated with ULK4 expression is associated with diastolic blood pressure and pulse pressure, among other examples (highlighted in Supplementary Table 13). Extending these analyses using more WGS data will allow the integration of more, and rarer, MEVs in GWAS of additional phenotypes, leading to the discovery of additional disease-causing MEs and motivating development of ME-targeting drugs. The observation that a human-specific ME insertion substantially predisposes to keloid, which has not been observed in other primates38, also supports the utility of this approach to infer genetic origins of other traits characteristic of our species39.
By improving detection and prioritization of a type of variants difficult to assess at genome-wide scale, our tool and results are applicable to medical genetics. Even so, a major limitation remains: confident prediction of which MEVs alter phenotype requires additional data integration and statistical testing. However, our results also demonstrate that ME ontology relates coherently to MEV effect. Here, we infer putative effects of several MEVs at the level of disease, providing important information for personalized medicine; MEVs impact many traits plausibly entangled with fitness in our varied landscapes, but we have not explicitly addressed beneficial variants or those with antagonistic pleiotropy. Still, our work provides comprehensive backing to the assertion that MEs are drivers of diversification of genome sequence and function, classic concepts of genome evolution. In addition, we highlight MEs as a source of biased mutation, invoked to account for neutral evolution of complexity40. As the direction and pace of diversification can be modified by MEs, differences in ME-derived mutation patterns may potentiate differential genome plasticity between lineages.
Overview of the algorithm of MEGAnE
MEGAnE finds ME insertions and absences and genotypes the discovered MEVs. It searches for discordantly mapped reads and finds potential breakpoints from clipped reads. It uses BLASTn to search for similarity between the overhangs of clipped reads and ME insertions. It makes breakpoint pairs that represent the upstream and downstream breakpoints of an ME insertion or absence, or, in most cases, the start and end positions of a target site duplication (TSD). It then extracts breakpoints that are highly likely to derive from ME insertions or absences and fits a Gaussian mixture model, which models homozygosity and heterozygosity of the input sample. Based on the modeled distribution, MEGAnE removes likely false positives. After discovering ME insertions and absences, it genotypes the polymorphic MEs based on the number of reads providing evidence of each breakpoint, evidence of breakpoint absence and read depth of the TSD. It outputs discovered ME insertions and absences in VCF format (Supplementary Fig. 1).
After MEV discovery and genotyping of multiple samples, MEGAnE can merge them to make a joint callset. It first merges the breakpoint positions in multiple VCF files, then searches for reads providing evidence of the merged breakpoints. If sufficient reads support a breakpoint, discrete genotypes (that is ‘0/1’ or ‘1/1’) are assigned. If there are no reads supporting a breakpoint, it assigns genotypes as ‘0/0’. If there is weak evidence of the breakpoint, it leaves the genotype as missing, that is ‘./0’.
MEV discovery from 1000GP GRCh38 datasets
The 30× WGS data from 3,202 individuals mapping to GRCh38DH were downloaded from the 1000GP website (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/). Throughout this paper, we refer to this dataset as ‘1000GP GRCh38 datasets’. MEs were discovered and genotyped using MEGAnE’s call_genotype_38 command. The joint callset was generated using MEGAnE’s joint_calling_hs command. We also generated a joint callset from 2,503 individuals, which does not include relatives, using from the same dataset. We generated a separate joint callset for 34 individuals who were sequenced using PacBio in the 1000GP HGSVC project. The HGSVC sequenced 35 individuals by PacBio; however, we excluded one individual, HG002, from our joint callset, because the individual was not included in the 3,202 individuals who were sequenced in the 1000GP 30× WGS. In 2,503 individuals analyzed here, MEGAnE detected 48,248 MEVs with the filter ‘PASS’ flag. Of those, 8,609 (18% of total) were common variants (AF > 1%).
MEV discovery from 1000GP GRCh37 datasets
The raw fastq reads of the 2,504 individuals in the 1000GP 30× GRCh38 datasets were downloaded from the 1000GP website (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/). The fastq reads were mapped on the human reference genome build, human_g1k_v37 by BWA MEM using the same options as used by 1000GP to map on GRCh38DH (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/20190405_NYGC_b38_pipeline_description.pdf). In brief, we used the -Y option with the -K 100000000 option. Throughout this paper, we refer to this dataset as ‘1000GP GRCh37 datasets’. The output alignment was converted to CRAM format and analyzed using MEGAnE’s call_genotype_37 command. The joint callset was generated using MEGAnE’s joint_calling_hs command. In 2,504 individuals analyzed here, MEGAnE detected 48,360 MEVs with the filter ‘PASS’ flag. Of those, 8,665 (18% of total) were common variants (AF > 1%) (Fig. 1e).
MEV discovery from 25× WGS datasets in BBJ
We applied MEGAnE to the 25× WGS (either 160 or 150 bp paired-end) from 1,235 individuals in BBJ41,42. We mapped the raw fastq reads to the human reference genome hs37d5 by BWA MEM using the same option as we used for mapping of 1000GP dataset and saved as CRAM format. We did not perform further individual-level QC, because the dataset was already subjected to QC. The output CRAM files were analyzed by the MEGAnE’s call_genotype_37 command. The joint callset was generated by the MEGAnE’s joint_calling_hs command. In 1,235 Japanese individuals analyzed here, it detected 10,996 MEVs with the filter ‘PASS’ flag. Of those, 4,943 (45% of total) were common variants (AF > 1%). This callset was used for evaluating LD between MEVs and SNVs.
MEV discovery from 25× and 15× WGS datasets in BBJ
To find rare insertions in Japanese individuals, we generated a joint callset by merging data from as many Japanese individuals as possible. To this end, we analyzed additional 30× WGS (either 125 or 124-nt paired-end) from 256 individuals and 15× WGS (150 bp paired-end) from 3,389 individuals by MEGAnE and merged with the MEVs detected from 1,235 individuals described above. When analyzing 15× WGS, we used the ‘-lowdep’ option of MEGAnE, which assumes non-Gaussian distributions of supporting read counts in heterozygous and homozygous insertions. In total, we merged MEVs from 4,480 Japanese individuals using the joint_calling_hs command. In 4,880 Japanese individuals analyzed here, MEGAnE detected 24,933 MEVs with the filter ‘PASS’ flag. Of those, 5,452 (22% of total) were common variants (AF > 1%) (Fig. 1e). This joint callset was used to investigate ME insertion preferences in Japanese.
Haplotype estimation for MEGAnE callset 1000GP GRCh38
First, we merged the MEI and ME absence callsets from MEGAnE. We used MEGAnE’s reshape_vcf command to merge these two callsets and remove multi-alleic ME variants. To estimate haplotypes of 2,503 individuals in 1000GP phase3, we merged the ME callset with SNVs. For quality control (QC), we first split the ME callset into individuals belonging to each of five superpopulations and evaluated Hardy-Weinberg equilibrium. Variants that violated Hardy-Weinberg equilibrium (P < 1 × 10−6) in at least one superpopulation were removed. SNVs that overlap with polymorphic MEs were removed. Singleton SNVs and MEs were also removed. Then, the QC-ed ME callset was merged with the SNV callset (1000GP, GRCh38_v1a) without variants violating Hardy-Weinberg equilibrium (P < 1 × 10−6). Each chromosome of the merged callset was saved in VCF format and phased using SHAPEIT4 software with default genetic maps. The phased haplotypes were converted to an imputation reference panel using Minimac3 software. Due to the unavailability of SNVs on sex chromosomes, we estimated the haplotypes for MEs only on autosomes and PARs.
Haplotype estimation for MEGAnE callset 1000GP GRCh37
First, we merged the MEI and ME absence callsets from MEGAnE using the same MEGAnE command described in the previous section. To estimate haplotypes of 2,504 individuals in 1000GP phase3, we merged the ME callset with SNVs and indels. We first split the ME callset into individuals belonging to each of five superpopulations and evaluated Hardy-Weinberg equilibrium. Variants that violated Hardy-Weinberg equilibrium (P < 1 × 10−6) in at least one superpopulation were removed. SNVs and indels that overlap with polymorphic MEs were removed. Singleton SNVs, indels, and MEs were also removed. Then, the QC-ed ME callset was merged with the SNV and indel callset (1000GP, v5a) without variants violating Hardy-Weinberg equilibrium (P < 1 × 10−6). Each chromosome of the merged callset was saved in VCF format and phased by SHAPEIT4 software with default genetic maps. An imputation reference panel was made using Minimac3 software. Due to the unavailability of SNVs on the Y chromosome, we estimated haplotypes for MEs only on autosomes and the X chromosome.
Genotype imputation for GTEx individuals
To impute ME genotypes in 838 individuals recruited in the GTEx v8, we used the 5,006 haplotypes in 1000GP. We used the phased SNVs and indels provided from GTEx (GTEx_Analysis_2017-06-05_v8_WholeGenomeSeq_838Indiv_Analysis_Freeze.SHAPEIT2_phased.vcf.gz) as target haplotypes. Variants violating Hardy-Weinberg equilibrium (P < 1 × 10−6) were removed before imputation. ME genotypes on autosomes and PARs were imputed using Minimac3 software with the imputation reference panel generated from the 1000GP GRCh38 callset. After imputation, ME genotypes were extracted and merged with the original SNV and indel calls. MEs violating Hardy-Weinberg equilibrium (P < 1 × 10−6) and/or having Minimac R2 lower than 0.5 were removed. Variants with allele frequency lower than 0.5% were removed, leaving 9,836 MEVs for use in eQTL analysis.
Genotype imputation in BBJ
To impute ME genotypes of participants in BBJ, we used the 5,008 haplotypes in the 1000GP GRCh37 dataset. We used phased SNVs genotyped by SNV array as target haplotypes. ME genotypes on autosomes were imputed using Minimac3 software with the imputation reference panel generated from the 1000GP GRCh37 callset. After imputation, variants violating Hardy-Weinberg equilibrium (P < 1 × 10−6) and those with Minimac R2 lower than 0.7 were removed. All variants with minor allele count lower than 10 were removed, and the remaining variants were used for GWAS.
PC analysis of MEVs
The PCs of ME polymorphisms called from 1000GP GRCh37 datasets and the SFARI cohort were calculated by Plink2 software. We first removed MEVs violating Hardy-Weinberg equilibrium (P < 1 × 10−6), those with minor allele frequency lower than 1%, and those in regions of long-range high LD (https://genome.sph.umich.edu/wiki/Regions_of_high_linkage_disequilibrium_(LD)). The variants were then pruned by Plink2 software with ‘–indep-pairwise 500 5 0.2’ option. The top 10 PCs were calculated using the plink2–pca command.
Intersections between MEVs and gene annotations
To compile MEVs that intersect with exons, CDS, and promoters, we first reshaped gene annotation files downloaded from GenCode using a script provided in the GTEx pipelines (https://github.com/broadinstitute/gtex-pipeline/blob/master/gene_model/collapse_annotation.py). We defined the 1-kb regions upstream from transcription start sites as promoters. All gene annotations in the GTF file were used for this analysis. To see intersection with MEVs called from 1000GP, we used 48,241 MEVs with the filter ‘PASS’ flag called from 1000GP GRCh38 datasets. For this analysis, we used a GenCode GTF version 26. To see intersection with MEVs called from BBJ, we used 10,997 MEVs with the filter ‘PASS’ flag called from 1,235 individuals sequenced at 25× depth WGS. For this analysis, we used a GenCode GTF version 26lift37.
Correlations between ME insertions and genomic features
To evaluate the characteristics of genome features found to have insertions of MEs, the correlation between the number of ME insertions and genomic features was calculated. We calculated the genomic features for nonoverlapping 100-kb windows (see the ‘Preparation of genomic features’ section). Because L1 and SVA insertions are sparse, we first resized the window size to 1 Mb and 5 Mb, respectively. To this end, the average values were calculated for each nonoverlapping window. Then, 1-Mb and 5-Mb windows that contain one or more 100-kb window(s) with missing value and ones with at least one ‘N’ character in the human genome assembly, GRCh38DH, were excluded from the analysis. The Spearman correlation coefficients were calculated using the SciPy module in Python.
eQTL analysis in 49 tissues
We performed eQTL mapping using MEVs. We followed the eQTL mapping method used in GTEx v8. As for GTEx v8, we excluded 5 tissues out of the 54 tissues (Bladder, Cervix_Ectocervix, Cervix_Endocervix, Fallopian_Tube, and Kidney_Medulla) from analysis due to the few available RNA-sequencing samples. First, expression profiles of the 49 tissues were prepared. The count per million matrices provided from GTEx (GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_reads.gct.gz) were normalized across samples by TMM normalization using the script provided from GTEx (https://github.com/broadinstitute/gtex-pipeline/blob/master/qtl/src/eqtl_prepare_expression.py), then the genes that are expressed (≥ 0.1 TPM in ≥ 20% samples and ≥6 reads in ≥20% samples) were retained for eQTL mapping (38,471 genes in total of 49 tissues). Each gene was then inverse-normal transformed across samples. Next, we performed eQTL mapping by fastQTL software43 with the same analysis options as for the previous eQTL mapping (https://github.com/broadinstitute/gtex-pipeline/tree/master/qtl). We also used the same covariates as those used for QTL mapping in GTEx: 5 genetic PCs, PEER factors, library preparation methods, sequencing platforms and sex. Genetic variants within 1 Mb from a gene were tested for associations. The 9,836 and 13,498,030 quality controlled ME and non-ME (that is SNVs and indels) variants, respectively, were used for eQTL mapping.
Across-tissue meta-analysis
After the eQTL mapping in each tissue, we performed across-tissue meta-analysis using the same method as performed in GTEx v8. First, we formatted the fastQTL results for MASH software44. Then, the MASH model was trained by the same protocol as GTEx v8 performed (https://github.com/stephenslab/gtexresults/blob/master/workflows/fastqtl_to_mash.ipynb). The trained model was applied to ME-eGene pairs.
Detection of ME-eQTL
We defined ME-eQTLs as those which satisfy these criteria: (1) in the fastQTL output, an MEV is either the lead variant or has r2 > 0.95 to the lead variant in at least one tissue, and (2) in the result of across-tissue meta-analysis, the MEV has local false sign rate < 0.05 in at least one tissue (Supplementary Table 9).
ME-GWAS of 42 diseases in BBJ
GWAS for 42 diseases were done using 179,660 individuals in BBJ using methods similar to those used in Ishigaki et al.45. The MEV genotypes in 179,660 individuals were imputed by Minimac3 software using the imputation reference panel generated from the 1000GP GRCh37 datasets. After imputation, variants violating Hardy-Weinberg equilibrium (P < 1 × 10−6), those with Minimac R2 lower than 0.7, and those with a minor allele count lower than 10 were removed. The associations were calculated using a generalized linear mixed model implemented in SAIGE (version 0.44.5)46 with the leave-one-chromosome-out approach. We used age, sex and the first five genetic PCs as covariates. For each disease, we defined a significantly associated locus as a genomic region within 3 Mb from the lead variants. Based on the methodology used in Ishigaki et al.45, we used 9.58 × 109 as a genome-wide significance threshold and 5 × 108 as a threshold of suggestive association.
Knockout of L1-NEDD4 in iPSCs
We designed two sgRNAs cleaving upstream and downstream of L1-NEDD4 insertion. To reconstruct the allele without the L1-NEDD4, we amplified the L1-flanking regions (703 bp upstream and 787 bp downstream) and connected them at the TSD using overlap-extension PCR. The connected fragment was used as a template for homology-directed repair. The sgRNA-Cas9 complex and homology-directed repair template DNA were transfected to iPSCs derived from a healthy Japanese individual (60 s, male) found to carry two copies of L1-NEDD4 by electroporation using the NEON transfection system. After electroporation, cells were cultured for 2 weeks, and single cell-derived clones were obtained by limiting dilution. Deletion of L1-NEDD4 was checked by the same primers as used for PCR validation in 70 Japanese (Supplementary Fig. 44b).
Differentiation of iPSCs into fibroblasts
iPSC clones were first differentiated to mesenchymal stem cells (MSCs) using STEMdiff Mesenchymal Progenitor Kit according to the manufacturer’s protocol. iPSC-derived MSCs were then differentiated to fibroblasts based on the protocol published in Lee et al.47. MSCs were cultured in DMEM containing 100 ng ml−1 CTGF, 50 ng/ml ascorbic acid, 1× penicillin/streptomycin, and 10% FBS for at least 3 weeks. Fully differentiated fibroblasts were maintained in the same medium used for MSC to fibroblast differentiation.
qRT-PCR of NEDD4 transcripts
To measure the expression levels of NEDD4 in fibroblasts, we collected L1-NEDD4 KO and WT clones differentiated into fibroblasts and extracted total RNA. Polyadenylated RNA was reverse-transcribed using oligo-dT primer. To measure the expression level of the long transcript variant of NEDD4, we designed primers in the long-variant-specific exons (exon 1 and 8). To measure expression of the short transcript variant of NEDD4, we designed primers amplifying the junction of the short-variant-specific exon (exon 9) and an exon that are shared in both short and long variants (exon 14), because exon 9 is the only exon that is specific to the short variant. Beta-actin transcript was used as an internal control. We also measured the expression of GAPDH, and the linearity between beta-actin and GAPDH expressions across samples was confirmed. The relative expression levels of the NEDD4 transcripts were calculated by ∆∆Ct method. We serially diluted cDNA to confirm that the qPCR conditions used resulted in exponential amplification. qPCR was performed on ViiA7 Real-Time PCR System using SYBR Green reagent. The sequences of the primers are listed in Supplementary Table 16.
Ethics approval
For all participating studies, we obtained informed consent from all participants by following the protocols approved by their institutional ethical committees. We obtained approval from the ethics committee of the RIKEN Center for Integrative Medical Sciences. We have complied with all the relevant ethical regulations.