David Adlam, Takiy-Eddine Berrandou, Adrien Georges, Christopher P. Nelson, Eleni Giannoulatou, Joséphine Henry, Lijiang Ma, Montgomery Blencowe, Tamiel N. Turley, Min-Lee Yang, Sandesh Chopade, Chris Finan, Peter S. Braund, Ines Sadeg-Sayoud, Siiri E. Iismaa, Matthew L. Kosel, Xiang Zhou, Stephen E. Hamby, Jenny Cheng, Lu Liu, Ingrid Tarr, David W. M. Muller, Valentina d’Escamard, Annette King, Liam R. Brunham, Ania A. Baranowska-Clarke, Stéphanie Debette, Philippe Amouyel, Jeffrey W. Olin, Snehal Patil, Stephanie E. Hesselson, Keerat Junday, Stavroula Kanoni, Krishna G. Aragam, Adam S. Butterworth, CARDIoGRAMPlusC4D, MEGASTROKE, International Stroke Genetics Consortium (ISGC) Intracranial Aneurysm Working Group, Marysia S. Tweet, Rajiv Gulati, Nicolas Combaret, DISCO register, Daniella Kadian-Dodov, Jonathan M. Kalman, Diane Fatkin, Aroon D. Hingorani, Jacqueline Saw, Tom R. Webb, Sharonne N. Hayes, Xia Yang, Santhi K. Ganesh, Timothy M. Olson, Jason C. Kovacic, Robert M. Graham, Nilesh J. Samani & Nabila Bouatia-Naji
7926 Accesses, 424 Altmetric
Abstract
Spontaneous coronary artery dissection (SCAD) is an understudied cause of myocardial infarction primarily affecting women. It is not known to what extent SCAD is genetically distinct from other cardiovascular diseases, including atherosclerotic coronary artery disease (CAD). Here we present a genome-wide association meta-analysis (1,917 cases and 9,292 controls) identifying 16 risk loci for SCAD. Integrative functional annotations prioritized genes that are likely to be regulated in vascular smooth muscle cells and artery fibroblasts and implicated in extracellular matrix biology. One locus containing the tissue factor gene F3, which is involved in blood coagulation cascade initiation, appears to be specific for SCAD risk. Several associated variants have diametrically opposite associations with CAD, suggesting that shared biological processes contribute to both diseases, but through different mechanisms. We also infer a causal role for high blood pressure in SCAD. Our findings provide novel pathophysiological insights involving arterial integrity and tissue-mediated coagulation in SCAD and set the stage for future specific therapeutics and preventions.
Main
Cardiovascular disease is the leading cause of death in women, but sex-specific aspects of the risk of heart disease and acute myocardial infarction (AMI) remain understudied1. Spontaneous coronary artery dissection (SCAD) and atherosclerotic coronary artery disease (CAD) are both causes of acute coronary syndromes leading to AMI2,3,4,5,6. However, in contrast with CAD, SCAD affects a younger, predominantly female population7 and arises from the development of a hematoma, leading to dissection of the coronary tunica media with the eventual formation of a false lumen, rather than atherosclerotic plaque erosion or rupture8. SCAD has been clinically associated with migraine9 and extra-coronary arteriopathies, including fibromuscular dysplasia. However, co-existent coronary atherosclerosis is uncommon8,14. While the genetic basis of CAD is increasingly well established15, the pathophysiology of SCAD remains poorly understood4. The search for highly penetrant mutations in candidate pathways or by sequencing has garnered a low yield, often pointing to genes involved in other clinically undiagnosed inherited syndromes manifesting as SCAD16. Previous investigations of the impact of common genetic variation on the risk of SCAD have described five confirmed risk loci.
In this Article, we performed a meta-analysis of genome-wide association studies (GWASs) comprising 1,917 SCAD cases and 9,292 controls of European ancestry. We identified 16 risk loci, including 11 new association signals, demonstrating a substantial polygenic heritability for this disease. Importantly, we show that several common genetic risk loci for SCAD are shared with CAD but have a directionally opposite effect and a different genetic contribution of established cardiovascular risk factors. These findings implicate arterial integrity related to extracellular matrix biology, vascular tone and tissue coagulation in the pathophysiology of SCAD.
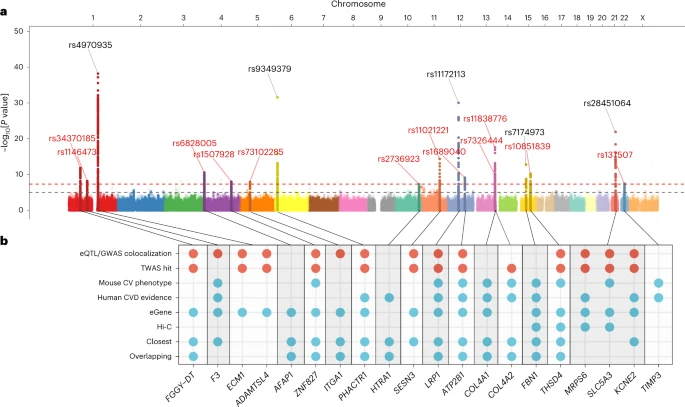
Results
GWAS meta-analysis and single-nucleotide polymorphism heritability. We conducted a GWAS meta-analysis of eight independent case–control studies. Sixteen loci demonstrated genome-wide-significant signals of association with SCAD, among which 11 were newly described for this disease. One locus on chromosome 4 (AFAP1) was recently reported for SCAD in the context of pregnancy19 and has now been confirmed as being generally involved in SCAD (Table 1). The estimated odds ratios of associated loci ranged from 1.25 (95% confidence interval (CI) = 1.16–1.35) in ZNF827 on chromosome 4 to 2.04 (95% CI = 1.77–2.35) on chromosome 21 near KCNE2 (Table 1). We report evidence for substantial polygenicity for SCAD with an estimated single-nucleotide polymorphism (SNP)-based heritability above 0.70 (h2SNP = 0.71 ± 0.11 on the liability scale using linkage disequilibrium score regression21 and h2SNP = 0.70 ± 0.12 using SumHer22; Supplementary Table 3). The ECM1/ADAMTSL4 locus on chromosome 1 accounted for the largest proportion of heritability for SCAD in our dataset (h2 = 0.028), followed by the COL4A1/COL4A2 locus, which contained two independent GWAS signals (h2 = 0.022; Supplementary Table 4 and Supplementary Fig. 4). Overall, we estimate that the 16 loci explain ∼24% of the total SNP-based heritability of SCAD (Supplementary Table 4).
Discussion
In this Article, we provide the largest study to date aimed at understanding the genetic basis of SCAD—an understudied cause of AMI that primarily affects women. We report novel associations and demonstrate high polygenic heritability for SCAD. We leverage integrative functional annotations to prioritize genes that are likely to be regulated in VSMCs and the fibroblasts of arteries. Insights from the biological functions of genes highlight the central role of extracellular matrix integrity and reveal impaired tissue coagulation as a novel potential mechanism for SCAD. Globally, we demonstrate the polygenic basis of SCAD to be shared with an important set of cardiovascular diseases. However, a striking directionally opposite genetic impact is found with atherosclerotic CAD, involving multiple risk loci and leading to a genome-wide negative genetic correlation. We provide evidence supporting genetically predicted higher blood pressure as an important risk factor for SCAD, but not other well-established cardiovascular factors. Our results set the stage for future investigation of novel biological pathways relevant to both SCAD and CAD and potential therapeutic and preventive strategies specifically targeting SCAD.
As an understudied condition that was previously thought to be uncommon, SCAD was initially suspected to involve rare and highly penetrant mutations. However, recent sequencing studies have suggested that only a small proportion (~3.5%) of SCAD cases are due to rare variants16,32. This is in keeping with increasing clinical recognition suggesting that this condition is not rare and occurs globally in populations of both European and non-European ancestry, with similar disease characteristics and probably similar prevalence2,4,33,34. Despite a modest sample size, we identified 16 risk loci accounting for about one-quarter of the polygenic heritability, which we estimate to be as high as ∼71%, therefore indicating that SCAD is predominantly a complex polygenic disease. However, we acknowledge that larger GWAS settings, including ancestrally diverse populations, will enhance the statistical power needed to provide validation through replication of the reported risk loci and estimated polygenic heritability.
This study supports the presence of genetic overlap between the risk of SCAD and other vascular diseases involving generally younger individuals and more women, such as cervical arterial dissection, migraine, subarachnoid hemorrhage and FMD. These conditions are reported to occur at increased frequency in patients with SCAD10,11,12,13, supporting shared causal biological mechanisms. Among the genes we prioritize as novel SCAD loci, we highlight the ATPase plasma membrane Ca2+ transporting 1 gene (ATP2B1) that we recently reported to associate with FMD35—a well-established locus for blood pressure risk36 via its role in intracellular calcium homeostasis in VSMCs and blood pressure regulation37. Most importantly, we provide evidence for a causal genetic effect of both SBP and DBP in SCAD risk. These findings provide an important genetic basis to support observational data suggesting that control of blood pressure may be an important factor in reducing the risk of recurrence after SCAD38. However, our findings also suggest that controlling other causal risk factors for CAD, such as LDL cholesterol with statins, may confer less benefit in SCAD than in CAD.
Knowledge of the molecular mechanisms leading to SCAD has been limited. Insights from sequencing studies of rare genetic variants have shown that most are associated with genes known from hereditary connective tissue disorders such as vascular Ehlers–Danlos, Loeys–Dietz and Marfan syndromes, as well as adult polycystic kidney disease16,32. A striking finding from our study is the identification of the tissue factor gene F3—a critical component of tissue-mediated blood coagulation—as a strong candidate gene in a risk locus for SCAD. We found that genetically determined lower expression of F3 in arterial tissue was associated with a higher risk for SCAD, involving variants located in putative functional regulatory elements in the coronary artery, VSMCs and fibroblasts. Tissue factor is synthesized at the subendothelial level of VSMCs and by fibroblasts in the adventitia surrounding the arteries39. In SCAD, once an intramural hemorrhage has initiated, propagation and pressurization of the false lumen may depend, in part, on coagulation and stabilization of the hematoma. Tissue factor is also a druggable target, albeit a potentially challenging one given its known multiple physiological and pathophysiological roles ranging from hemostasis to cancer metastasis. Tissue factor is widely studied in the context of prothrombotic conditions, including atherosclerosis, although notably the genetic variants we describe here do not associate with atherosclerotic disease. This feature is an exception to the highly pleiotropic nature of the variants we describe in the remaining SCAD loci, suggesting impaired tissue-initiated coagulation as a putative specific mechanism in SCAD.
We identify regulation of the extracellular matrix of arteries as the predominant polygenic biological mechanism for SCAD. Integrative prioritization analyses revealed 13 potential causal genes with established key roles in maintaining arterial wall integrity and function. Among these, we highlight the serine protease HTRA1 and metallopeptidase inhibitor TIMP3, which are involved in matrix disassembly. TIMP3 clusters in the main network for extracellular matrix organization that includes ADAMTSL4, LRP1 and COL4A1, with connections with subnetworks of F3. This clustering is consistent with the biological function of TIMP3 as an inhibitor of matrix metalloproteinases with domains interacting with ADAMTS proteins and LRP1, involving proteins encoded by genes prioritized in SCAD loci40. Interestingly, we found a novel association signal with SCAD in the metallopeptidase thrombospondin type 1 domain containing 4 gene (THSD4) that promotes fibrillin 1 elastic fiber assembly, and confirm the previously reported associations near ADAMTSL4 and FBN1 (refs. 18,20). We showed that genetically decreased expressions of these genes in arteries were correlated with higher SCAD risk alleles in arteries or fibroblasts. This finding suggests that a genetic predisposition to a weaker extracellular matrix may increase the vulnerability of traversing intramural microvessels to disruption, increasing the risk of initiation and propagation of a false lumen within the coronary vessel wall, leading to SCAD.
Many of the risk loci for SCAD that we report here, as well as their prioritized genes, are already known from atherosclerotic disease GWASs. However, here we provide compelling and intriguing evidence for the opposite directionality of a substantial fraction of genetic bases for SCAD versus CAD, suggesting that some key biological mechanisms involved in the two diseases are also likely to be opposite, which is consistent with the clinical observation of a lower-than-expected burden of atherosclerotic disease in patients with SCAD. For example, the association signals in the COL4A1/COL4A2 locus are in an opposite direction to their contribution to CAD41. This locus encodes α1 and α2 chains of type IV collagen, with transcripts generated through a common promoter. Type IV collagen is the main component of the basement membrane of arterial cells and plays a key role in the structural integrity and biological functions of VSMCs in the tunica muscularis. Decreased collagen IV expression increases the risk of CAD15,42. Proposed potential mechanisms for this include a disinhibition of VSMC-intimal migration during atherogenesis or an increase in the vulnerability of atherosclerotic plaque to rupture42. In contrast with CAD, our data indicate that genetically mediated increased collagen IV expression also increases the risk of SCAD. Better understanding of how these directionally opposite changes modify the risk of CAD and SCAD has considerable potential to enhance our understanding of the molecular genetic mechanisms that confer risk in both diseases.