Veera M. Rajagopal, Kyoko Watanabe, Joelle Mbatchou, Ariane Ayer, Peter Quon, Deepika Sharma, Michael D. Kessler, Kavita Praveen, Sahar Gelfman, Neelroop Parikshak, Jacqueline M. Otto, Suying Bao, Shek Man Chim, Elias Pavlopoulos, Andreja Avbersek, Manav Kapoor, Esteban Chen, Marcus B. Jones, Michelle Leblanc, Jonathan Emberson, Rory Collins, Jason Torres, Pablo Kuri Morales, Roberto Tapia-Conyer, GHS-REGN DiscovEHR collaboration, Regeneron Genetics Center, Giovanni Coppola
8900 Accesses, 1 Citations, 7 Altmetric
Abstract
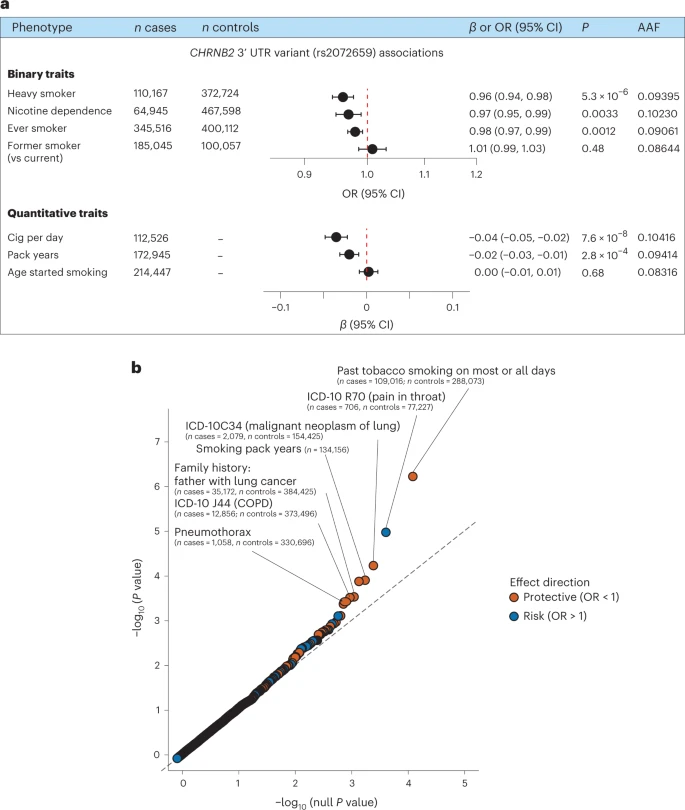
Human genetic studies of smoking behavior have been thus far largely limited to common variants. Studying rare coding variants has the potential to identify drug targets. We performed an exome-wide association study of smoking phenotypes in up to 749,459 individuals and discovered a protective association in CHRNB2, encoding the β2 subunit of the α4β2 nicotine acetylcholine receptor. Rare predicted loss-of-function and likely deleterious missense variants in CHRNB2 in aggregate were associated with a 35% decreased odds for smoking heavily (odds ratio (OR) = 0.65, confidence interval (CI) = 0.56–0.76, P = 1.9 × 10−8). An independent common variant association in the protective direction (rs2072659; OR = 0.96; CI = 0.94–0.98; P = 5.3 × 10−6) was also evident, suggesting an allelic series. Our findings in humans align with decades-old experimental observations in mice that β2 loss abolishes nicotine-mediated neuronal responses and attenuates nicotine self-administration. Our genetic discovery will inspire future drug designs targeting CHRNB2 in the brain for the treatment of nicotine addiction.
Results
Exome-wide significant associations.
The overall study design is shown in Fig. 1. We performed ExWAS meta-analyses for six primary phenotypes (ever smoker, heavy smoker, former smoker, nicotine dependence, cigarettes smoked per day (cig per day) and age started smoking) in sample sizes ranging from 112,670 (cig per day) to 749,459 (ever smoker). The study cohorts and phenotype definitions are described in the Methods, and the cohort-specific sample sizes and participant demographics are summarized in Supplementary Tables 1 and 2, respectively. We focused on coding variants of two functional categories: missense variants and predicted loss-of-function (pLOF) variants (frameshift, splice donor, splice acceptor, stop lost, stop gain and start lost) with MAF < 0.01. In addition to variant-level associations, we also studied gene-level associations, using burden tests in which either pLOF variants only or pLOF and likely deleterious missense variants (that is, predicted to be deleterious by five different algorithms) in a gene are aggregated to create burden masks (or variant sets), which are then tested for association with the phenotypes (Methods)21. The burden masks were created using variants at five MAF thresholds (<0.01, <0.001, <0.0001, <0.00001 and singletons) (Supplementary Table 3). Altogether, we performed 8,417,987 association tests across six smoking phenotypes. Applying a false detection rate (FDR) of 1% (corresponding P value = 4.5 × 10−8), we identified 35 significant associations implicating three genes: ASXL1, DNMT3A and CHRNB2 (Fig. 2, Supplementary Fig. 1 and Supplementary Table 4). Although these results were based on analyses in which individuals of all ancestries were pooled together, we found that the results were highly similar to those from a cross-ancestry meta-analysis or a meta-analysis involving only individuals of European ancestry, suggesting that the results were not influenced by population stratification.