Andras Sziraki, Ziyu Lu, Jasper Lee, Gabor Banyai, Sonya Anderson, Abdulraouf Abdulraouf, Eli Metzner, Andrew Liao, Jason Banfelder, Alexander Epstein, Chloe Schaefer, Zihan Xu, Zehao Zhang, Li Gan, Peter T. Nelson, Wei Zhou & Junyue Cao
13k Accesses, 122 Altmetric
Abstract
Conventional methods fall short in unraveling the dynamics of rare cell types related to aging and diseases. Here we introduce EasySci, an advanced single-cell combinatorial indexing strategy for exploring age-dependent cellular dynamics in the mammalian brain. Profiling approximately 1.5 million single-cell transcriptomes and 400,000 chromatin accessibility profiles across diverse mouse brains, we identified over 300 cell subtypes, uncovering their molecular characteristics and spatial locations. This comprehensive view elucidates rare cell types expanded or depleted upon aging. We also investigated cell-type-specific responses to genetic alterations linked to Alzheimer’s disease, identifying associated rare cell types. Additionally, by profiling 118,240 human brain single-cell transcriptomes, we discerned cell- and region-specific transcriptomic changes tied to Alzheimer’s pathogenesis. In conclusion, this research offers a valuable resource for probing cell-type-specific dynamics in both normal and pathological aging.
Main
Progressive changes in brain cell populations, which can occur during aging, may contribute to functional decline and increased risks for neurodegenerative diseases such as Alzheimer’s disease (AD)1,2,3,4. Although the recent advances in single-cell genomics have created unprecedented opportunities to explore the cell-type-specific dynamics across the entire mammalian brain5,6,7,8, most prior studies relied on a relatively shallow sampling of the brain cell populations and failed to reveal rare aging or AD-associated cell types. Additionally, they were technically limited in several ways, including failing to recover isoform-level gene expression patterns and the associated chromatin landscape that regulates cell-type-specific alterations across aging stages.
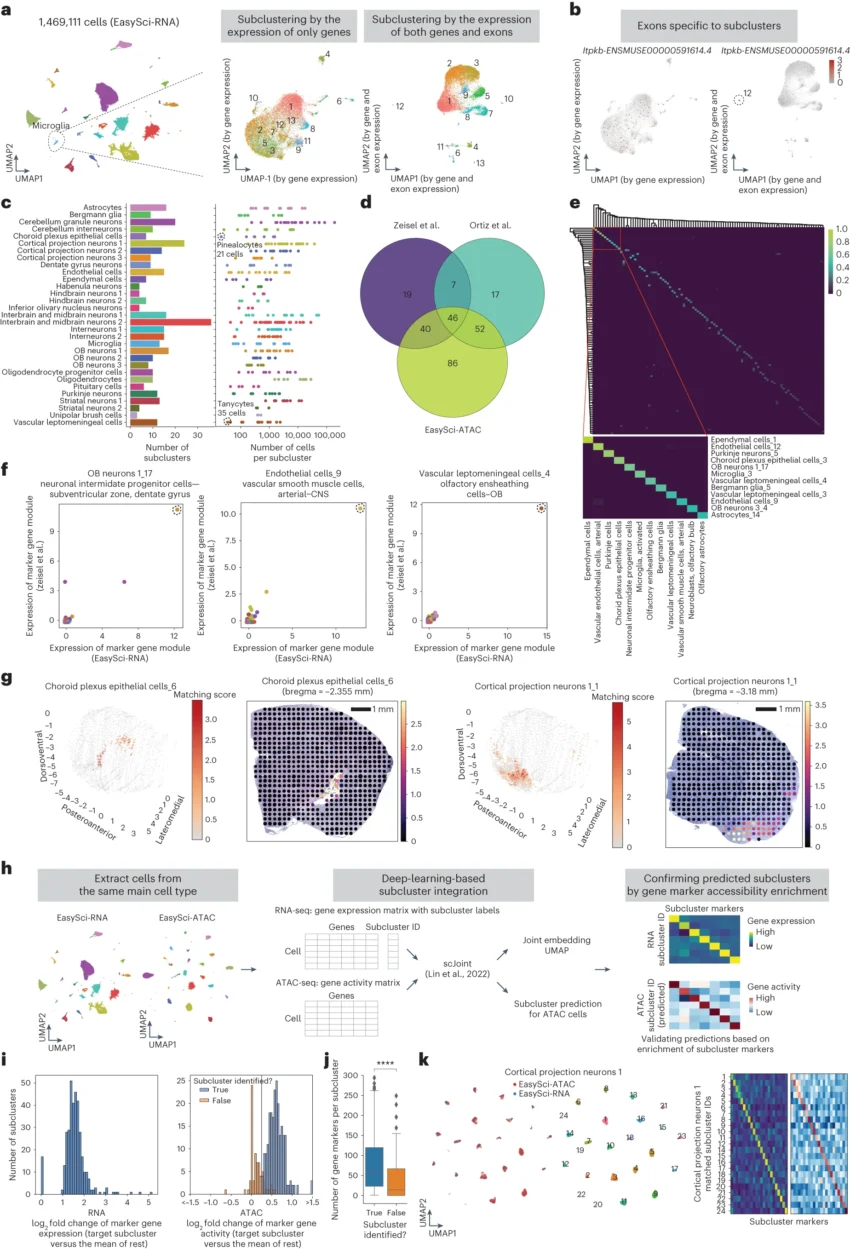
Here, we introduced EasySci, a cost-effective single-cell profiling strategy based on extensive optimization of single-cell RNA sequencing (RNA-seq) by combinatorial indexing. While the original method has been widely used to study embryonic and fetal tissues, it remains restricted to gene quantification proximal to the 3’ end and limited in efficiency and cell recovery rate. EasySci provided improved conditions for cell lysis, fixation, sample preservation, enzymatic reaction, oligonucleotide design, and purification methodologies. Several test conditions were inspired by optimizations described in recently developed or optimized single-cell techniques. The major features of EasySci include (i) 1 million single-cell transcriptomes were prepared for ~US $700 (library preparation cost only, not including personnel or sequencing cost; (ii) reverse transcription (RT) with indexed oligo-dT and random hexamer primers was achieved, thus recovering cell-type-specific gene expression with full gene body coverage; (iii) cell recovery rate, as well as the number of transcripts detected per cell, were substantially improved through optimized nuclei storage, enzymatic reactions and improved primer design; and (iv) an extensively improved single-cell data processing pipeline was developed for both gene counting and exonic counting using paired-end single-cell RNA-seq data.
Discussion
In this study, we introduced EasySci, a cost-effective technical framework for individual laboratories to generate gene expression and chromatin accessibility profiles from millions of single cells. We used EasySci to analyze 1.5 million single-cell transcriptomes with full gene body coverage and 380,000 chromatin accessibility profiles across mammalian brains of different ages and genotypes. The datasets enable the identification of over 300 cellular subtypes throughout the brain, including highly rare cell types representing less than 0.01% of the total brain cell population. Furthermore, we discovered region-specific effects attributable to aging and AD and examined the manifestation of molecular signatures associated with aging and AD on a cell-type-specific basis.
As highlighted by our subcluster level analysis, the effects of aging and AD on the global brain cell population are profoundly cell-type specific. Although most brain cell types stay relatively stable under various conditions, we identified over 50 cell subtypes exhibiting over twofold change in brains affected by aging and AD models. Many of these cell subtypes were rare and overlooked in conventional single-cell analysis. For example, the aging brain is characterized by the depletion of both rare neuronal progenitor cells and differentiating oligodendrocytes, associated with the enrichment of a C4b+ Serpina3n+ reactive oligodendrocyte subtype surrounding the SVZ, suggesting a potential interplay between oligodendrocytes, localized inflammatory signals and the stem cell niche.
The lack of reliable mouse models remains a big challenge in studying late-onset AD. The novel APOE*4/Trem2*R47H model aims to overcome this limitation by introducing two of the strongest late-onset AD-associated mutations. We found consistent molecular and cellular population dynamics between the well-established 5xFAD and the novel APOE*4/Trem2*R47H model. For example, we observed shared subtypes that were depleted (for example, mt-Cytb+ mt-Rnr2+ choroid plexus epithelial cell) or enriched (for example, Col25a1+ Ndrg1+ interbrain and midbrain neuron) in both early- and late-onset AD mutant brains. Meanwhile, differences were also observed between the two AD models, as expected by the different onset times. The absence of an increase in the DAM population in the LOAD model may be due to its lack of amyloid deposition or by genetic perturbations, as both Trem2 and Apoe play a role in the activation of this cell population.
In addition, we investigated AD-associated gene signatures in human brains by profiling over 100,000 single-nucleus transcriptomes derived from 24 human brain samples from control and AD patients, across two distinct anatomical locations. Although most AD-associated gene dynamics are profoundly cell-type and region specific, we identified dysregulated genetic signatures that are conserved between different locations in the human brains. Moreover, integrating the human and mouse brain datasets further revealed molecular pathways shared between human AD patients and mouse AD models, which suggests that the mouse AD model can serve as a model system to investigate the function and regulation of these conserved features associated with AD or neuronal dysfunction.
Of note, there are several inherent limitations of the study. First, the analysis covers only around 2% of the total mouse brain population (estimated at approximately 100 million cells), which means extremely rare cell subtypes may still be overlooked. Additionally, our relatively shallow sequencing depth might hinder the detection of lowly expressed transcripts or minor aging-related cellular state changes. Nevertheless, the validity of our key biological findings is reinforced by the consistent results across different genders (male versus female), genotypes (EOAD versus LOAD), and orthogonal approaches (such as comparisons between single-cell transcriptome, chromatin accessibility or spatial transcriptomics). This lends significant credence to our discoveries, even when considering the limitations of the study.
In summary, we have showcased the power of highly scalable single-cell genomics to delve into the dynamics of rare cell types, uncovering novel subtypes associated with aging and disease. Though our focus was on brain tissues, the strategic approach could be readily extended to systematically explore cellular states across an entire organism. Such exploration could illuminate the rare vulnerable cell populations to aging and diseases, opening up pathways to develop targeted therapeutic strategies.