Robin J. Hofmeister, Diogo M. Ribeiro, Simone Rubinacci & Olivier Delaneau
3309 Accesses, 68 Altmetric
Abstract
Phasing involves distinguishing the two parentally inherited copies of each chromosome into haplotypes. Here, we introduce SHAPEIT5, a new phasing method that quickly and accurately processes large sequencing datasets and applied it to UK Biobank (UKB) whole-genome and whole-exome sequencing data. We demonstrate that SHAPEIT5 phases rare variants with low switch error rates of below 5% for variants present in just 1 sample out of 100,000. Furthermore, we outline a method for phasing singletons, which, although less precise, constitutes an important step towards future developments. We then demonstrate that the use of UKB as a reference panel improves the accuracy of genotype imputation, which is even more pronounced when phased with SHAPEIT5 compared with other methods. Finally, we screen the UKB data for loss-of-function compound heterozygous events and identify 549 genes where both gene copies are knocked out. These genes complement current knowledge of gene essentiality in the human genome.
Main
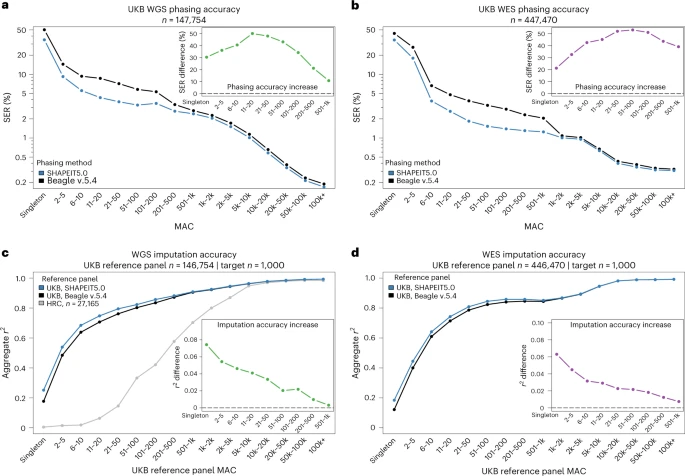
Modern genetic association studies are increasingly based on whole-genome or whole-exome sequencing (WGS/WES) for hundreds of thousands of samples collected as part of nationwide biobanking initiatives1,2. Compared with previous studies based on single nucleotide polymorphism (SNP) arrays, WGS and WES data can identify rare variants (e.g., minor allele frequency below 1%), allowing a systematic characterization of their contribution to trait heritability3, functional relevance4 and effects on various traits and diseases5,6. In this context, haplotype phasing of rare variants, which involves distinguishing the two parentally inherited copies of each chromosome into haplotypes, adds a layer of biologically relevant information and unlocks new analyses. For instance, phasing is crucial to identify compound heterozygous events, which occur when both copies of a gene contain nonidentical, heterozygous mutations. In the case of Mendelian disorders, compound heterozygosity is one of the most common inheritance models for rare recessive diseases in nonconsanguineous individuals7,8. Previous efforts to identify compound heterozygous events in large cohorts provided valuable insights, yet these either relied on imputed data9 or ignored phasing information6. Compound heterozygous event identification requires high-confidence phase information to be considered when rare variants are analyzed, such as in gene-based burden test analysis10. The most common approach to phase rare variants without parental genomes or long-reads in large cohorts of individuals is statistical phasing, which leverages information across individuals to make estimation of haplotypes11. This technique is well established for common variants typed on SNP arrays, where phase information is used, for instance, to perform genotype imputation12, admixture analysis13 and genealogy estimation14. Phasing methods have been optimized to scale to the thousands of samples in modern SNP array datasets, and the time is ripe to do the same for the millions of rare variant sites present in WGS/WES datasets. As an example, the WGS data for 150,119 UKB samples comprise three orders of magnitude more variants than the Axiom array data, around 96% of them having a minor allele frequency (MAF) below 0.1%. Phasing large scale WGS/WES datasets is challenging and new methods able to handle large amounts of rare variants are now emerging15. Recently, a computationally efficient solution for rare variant phasing has been implemented in Beagle v.5.4 (refs. 16,17), in which common and rare variants are phased separately: in a first step, a standard phasing method is used to obtain haplotypes at common variants, and in a second step rare heterozygous sites are phased onto the resulting haplotypes using genotype imputation technique. This type of strategy, based on haplotype scaffolds, has been used in other contexts, such as in genotype imputation18, integration of family data19 and external phasing information20.
In this work, we describe SHAPEIT5, a method designed to accurately phase rare variants in large WGS/WES datasets, including singletons, with moderate accuracy, while attributing phasing confidence scores. We applied it to estimate haplotypes for 150,119 and 452,644 UKB samples with WGS and WES data, respectively. We demonstrate the benefit of using these two haplotype collections as reference panels for SNP array imputation and finally show that the phase inferred at rare variants in the WES dataset can be screened to reliably identify compound heterozygous loss-of-function (LoF) mutations, probably leading to complete gene knockouts.
Results
Overview of the SHAPEIT5 phasing method
SHAPEIT5 performs haplotype phasing of WGS or WES data using three different phasing models, each focusing on a specific type of variants: (1) common variants are phased using the SHAPEIT4 model20, (2) rare variants are phased onto the resulting haplotypes using an imputation model and (3) singletons are phased using a coalescent-inspired model. See Fig. 1 for an illustration of the phasing scheme. Common variants are defined as having a MAF above 0.1% and are phased using an optimized version of the SHAPEIT4 algorithm, known to perform well on large sample sizes.
Discussion
We present SHAPEIT5, a tool for phasing rare variants in large sequencing datasets. SHAPEIT5 phases common variants first to create a haplotype scaffold. Subsequently, rare variants are phased one at a time on this scaffold. A key difference from Beagle v.5.4 is the use of individualized panels of haplotypes for rare variant phasing. SHAPEIT5 ensures representation of the minor alleles at rare variants, which leads to accuracy improvements that are more pronounced in larger sample sizes. We produced phased genomes for the UKB WGS and WES data for a compute cost below £4,000. The haplotype estimates have low SERs, with rare variants down to doubletons being phased with high confidence. This accurate phasing enables highly accurate genotype imputation when used as a reference panel. Beyond measuring error rates, we also validated phased haplotypes biologically by identifying compound heterozygous events, which we found highly depleted in essential genes, as expected. In addition, we achieved singleton phasing, albeit with higher error rates and therefore with limited downstream utility. However, we view this as an advance in phasing models as previous approaches were unable to phase singletons.
Although of substantial interest, previous knowledge of compound heterozygous cases comes mostly from case studies in families7,8 and there is currently no method to identify these events in large biobanks systematically. Here, we show that high-quality phasing of rare variants with SHAPEIT5 allows compound heterozygosity to be studied at the biobank-scale level, which can greatly increase the number of events characterized compared with the use of family data, in addition to exploring their association with new phenotypes. As a proof-of-principle, we screened all protein-coding genes for compound heterozygous events with high-confidence LoF variants and found 549 genes predicted to be fully knocked out across 816 UKB individuals out of the 374,826 individuals considered in this study. This complements other lists of nonessential genes33, with the main difference that these knockouts are found in vivo in humans. Approximately 0.22% of the UKB cohort had at least one gene knockout by compound LoF heterozygous events. This observed frequency of events matches previous estimates in outbred healthy cohorts34. UKB participants are not expected to have any rare and/or severe genetic diseases as their average age is 56 years, which is after the age of onset for most rare diseases. This partially explains why the gene knockouts observed are strongly depleted in several lists of essential genes. However, we still found 52 genes deemed as essential in at least one of the essential gene lists we analyzed. We can conceive three possible scenarios to explain these specific cases. First, the mutations had a moderate impact on the individual and did not result in severe disease. As an example, we found one individual with pulmonary embolism while having a knockout of the essential gene ADAM19—a gene reported for its involvement in pulmonary disease35,36. Second, compensatory mutations can rescue the deleterious effect of the knockout. For instance, we observed one individual with a knockout of CFFTR—an essential gene found to be rescued by several gain-of-function mutations across the genome37,38,39. Finally, some of the compound heterozygous events discovered may be false positives driven by incorrect phasing or erroneous LoF annotations.
We foresee that rare variant phasing in large sequencing studies such as the UKB has the potential to unlock many applications and analyses. First, other types of functional variants can be screened for compound heterozygous effects, for instance, combining LoF and missense or regulatory variants40. Second, phase information can be included in rare variant burden testing approaches, which usually consider only a mixture of the two haplotypes. Third, using accurately phased reference panels allows phasing of extremely rare variants with high accuracy, even singletons to some extent, for any new sequenced genome from the same population. This is beneficial for diagnosis of rare and severe diseases caused by compound heterozygous effects, such as in the Genomics England dataset41, in which diagnosis yield could be increased by incorporating phase information.