Elizabeth T. Cirulli, Simon White, Robert W. Read, Gai Elhanan, William J. Metcalf, Francisco Tanudjaja, Donna M. Fath, Efren Sandoval, Magnus Isaksson, Karen A. Schlauch, Joseph J. Grzymski, James T. Lu & Nicole L. Washington
30k Accesses, 68 Citations, 68 Altmetric
Abstract
Understanding the impact of rare variants is essential to understanding human health. We analyze rare (MAF < 0.1%) variants against 4264 phenotypes in 49,960 exome-sequenced individuals from the UK Biobank and 1934 phenotypes (1821 overlapping with UK Biobank) in 21,866 members of the Healthy Nevada Project (HNP) cohort who underwent Exome + sequencing at Helix. After using our rare-variant-tailored methodology to reduce test statistic inflation, we identify 64 statistically significant gene-based associations in our meta-analysis of the two cohorts and 37 for phenotypes available in only one cohort. Singletons make significant contributions to our results, and the vast majority of the associations could not have been identified with a genotyping chip. Our results are available for interactive browsing in a webapp (https://ukb.research.helix.com). This comprehensive analysis illustrates the biological value of large, deeply phenotyped cohorts of unselected populations coupled with NGS data.
Introduction
Over the past decade, we have witnessed the growing depth and breadth of genome-wide association studies (GWAS) leveraging genotyped common variants. It has been shown that the most useful and predictive insights about the genetic effects of common variants only begin to appear as sample sizes reach into the hundreds of thousands. Modern resources like the UK Biobank (UKB, www.ukbiobank.ac.uk) that make thousands of phenotypes available to match these genetic data are proving a boon to our understanding of human genetics. In addition to identifying specific variants associated with traits, modern GWAS show that polygenic scores utilizing thousands of common variants together can explain a sizable portion of phenotypic variation and that genetic risk for one phenotype can help explain variation in another1,2,3.
Until now, the insights stemming from these large sample sizes have only been available for common and low frequency variants, with comprehensive studies not available below a minor allele frequency (MAF) of about 0.1%. It has been shown that as allele frequencies drop, the effect sizes of associated variants can increase beyond the limits imposed by natural selection on more common variants. In rare disease and family-based studies, aggregating phenotypically-similar probands to identify associated groups of rare variants has been crucial to our understanding of disease; exome and genome-based approaches are now standard of care for evaluating these patients. However, the impact of rare variants on common traits and sub-clinical phenotypes has only been examined for selected phenotypes as large exome and phenotypic datasets have not been available.
The release by the UKB of 49,960 exomes matched to thousands of phenotypes enables, for the first time, large-scale clinicalomics discovery, including analyses of rare variants, at scale14. We have coupled these data to the exomes of 21,866 participants in the Healthy Nevada Project (HNP, Renown Health, Reno, Nevada) who consented to research involving their electronic medical records.
Population-based analyses to identify statistically significant associations between traits and rare variants require a different methodology from the common variant methods to which the field has grown accustomed. Because the power to identify statistically significant rare variant associations decreases as the MAF decreases, discovery analyses require that these rare variants be grouped together in some way. The most common method to group rare variants together in population-based genetic analyses is at the level of the gene, usually via a collapsing test, combined multivariate and collapsing test, the sequence kernel association test (SKAT), or unified tests that can consider both a burden and nonburden situation (SKAT-O). Analysis methods that group rare variants have been used in exome and genome sequencing studies to successfully discover genes associated with many traits, such as myocardial infarction, amyotrophic lateral sclerosis, and blood pressure.
Results
Description of included datasets.
We used two cohorts in this analysis (Table 1). The first cohort was the set of sequenced UKB exomes. The UKB participants are between the ages of 40 and 69, and each has been extensively phenotyped, including consenting to making their medical records available. As described previously, the exome-sequenced set of UKB samples is enriched for individuals with MRI data, enhanced baseline measurements, hospital episode statistics, and linked primary care records (described for Category 170 at http://biobank.ctsu.ox.ac.uk/crystal/label.cgi?id=170). Of the 49,960 exome-sequenced individuals, 55% are female, and 40,468 are classified by the UKB as genetically of European ancestry (field 22006).
The second cohort included the exomes of 21,866 participants from the HNP. These are unselected patients from Northern Nevada (Renown Health, Reno, Nevada) who consented to research involving their electronic medical records. The participants in this cohort are aged 18–89+, and 68% are female. We classified 17,238 of these as European ancestry using principal component analysis based on 184,445 representative common variants (see “Methods”).
Collapsing rare variants
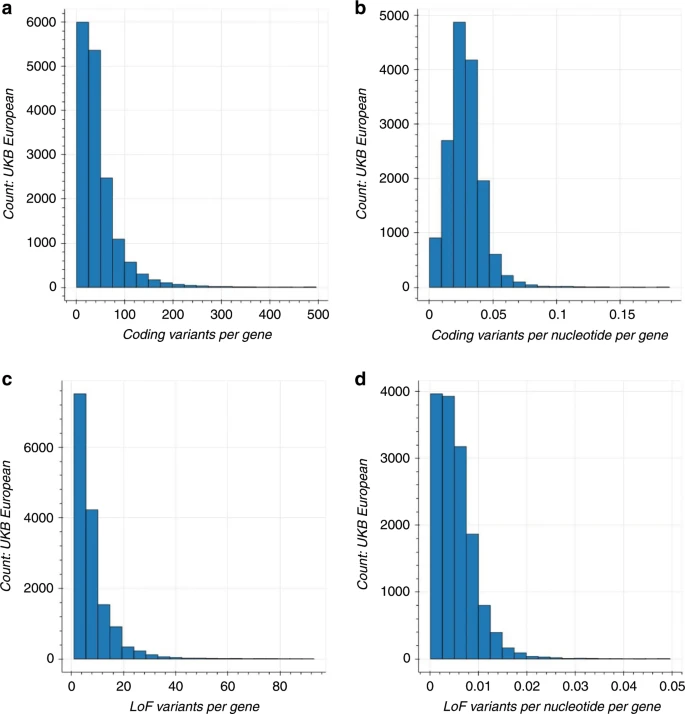
We performed a gene-based collapsing analysis to identify genes in which rare variants were, in aggregate, associated with a phenotype. In brief, we identified qualifying variants that met specific annotation criteria (see “Methods”) and had a MAF < 0.1%. We explored two gene-based collapsing models: (1) all non-benign coding and (2) only loss of function (LoF). The LoF model was used to identify associations where only putative LoF variants had an effect. In the coding model, we included 1,074,012 qualifying variants across 16,341 genes in the UKB cohort and 754,459 variants across 17,023 genes in the HNP cohort (see “Methods”). In the LoF model, we included 165,480 qualifying variants across 15,276 genes in the UKB cohort and 111,735 variants across 14,848 genes in the HNP cohort. There were 15,999 coding model genes and 13,474 LoF model genes that overlapped between the two cohorts. The median number of qualifying coding and LoF variants per gene in the UKB European ancestry population was 34 and six, and 22 coding and four LoF for the HNP cohort, respectively. When looking at all ethnicities, these numbers rose to 48 coding and seven LoF for the UKB cohort and 30 coding and seven LoF for the HNP cohort. The median percentage of people carrying qualifying variants in each gene was the same for both the UKB and HNP cohorts, both in European ancestry and across ethnicities: 0.13% for the coding model, and 0.02% for the LoF model.
Discussion
Here we present an analysis to catalogue the effects of rare and unique coding variants on thousands of phenotypes across two large cohorts. Until now, rare variant analyses using next generation sequence data have been performed on a small number of phenotypes at a time such as in schizophrenia, developmental delay, and diabetes33,34,35. Most of these studies were designed around specific phenotypes and collected targeted, disease-specific samples. Analyzing thousands of traits in a biobank-scale population presents additional challenges due to the rarity of the variants, which can lead to false positive associations. The best practices that we suggest to produce reliable results include restricting to high quality regions of the genome, setting a very low MAF cutoff, and requiring that at least a minimum threshold of individuals carry qualifying variants in the analyzed gene. Our methodology reduces test statistic inflation, and our success is demonstrated by the independent replication of most results.
Our analysis found that the majority of statistically significant gene-based associations were not driven by single explanatory variants. In fact, the signal for these associations was sufficiently dispersed over multiple variants per gene such that if aggregation at the gene-level was not utilized, 84% of the associated genes would not have had a single variant that exceeded the threshold for a multiple-testing correction. Furthermore, when the association for each gene was performed conditional on the single most significant variant in each gene, the p value for the gene as a whole remained below 5 × 10−8 for 85% of the associations.
Importantly, the associations identified in this analysis can only be obtained using sequencing techniques, as opposed to chip-based methods. All of the variants used in our analysis have a MAF below 0.1%, which is below the range of frequencies that can currently be comfortably imputed. Furthermore, 38% of the variants included in our analysis were singletons–only observed once in our dataset and never reported in gnomAD. Such unique variants are not accessible by chip and were vital to our study’s success. In fact, 88% of our statistically significant associations generated higher, i.e., worse, p values–by a median of 2 orders of magnitude–when the singletons were removed from the analysis.
In genes where multiple rare variants contribute to the signal, we find that mapping the precise contributions of each variant in the context of the secondary and tertiary structures can reveal the most functional parts of the gene for the given phenotype and provide additional support for a statistical association. Mapping individual missense variants to their sequence context after a gene-based discovery can also refine the classification of missense variants as true LoF. Corresponding diagrams mapping the locations of the rare variants in each significant gene-phenotype association can be found in Supplementary. Formal statistical tests of domain enrichment and discovery analyses that focus on different gene regions will doubtless uncover novel associations but will also often have less power due to the small number of people who will be carrying rare variants in each domain.
Our analysis differs from the one presented by Van Hout et al., who performed a gene-based analysis on the European ancestry subset of this same UKB dataset and with some of the same phenotypes. Some of our analysis differences included our use of a replication dataset, our more stringent MAF cutoff (1% vs. 0.1%), and collapsing model differences (LoF vs. both LoF and coding models). We also identified 5 of the 25 gene-based associations reported by Van Hout et al. (between TUBB1, IQGAP2, and KLF1 and blood cell phenotypes). The associations that we did not confirm were largely due to differences in our analysis techniques: the p value cutoff (three associations), the MAF cutoff (five associations), the requirement for a minimum number of carriers (six associations, none with p values via Fisher’s exact test that met our threshold), and restriction to high-confidence regions (three associations) (see Supplementary Table 1 for details). While the reasons for many of these study differences are innocuous, the associations with too few variant carriers in particular can be prone to false signals and will require larger sample sizes to confirm.
Many of the significant signals that we find reflect associations that had already been established in rare familial diseases (see Supplementary Data 2 for details). For example, we show that rare coding variants in GP1BB are associated with higher mean platelet volumes; previous studies have shown that variants in this gene cause certain familial bleeding and platelet disorders. Going beyond the acknowledgment that rare variants in this gene can cause these rare conditions, our findings contextualize what it means for any person who carries a non-benign variant in this gene: our work shows more broadly how rare variants in this gene may manifest sub-clinical blood-related phenotypes in the general population. This method brings us closer to a future where a single comprehensive calculation (incorporating both common and rare variation) is able to more accurately predict phenotypic outcomes of polygenic variation towards an improvement in our understanding of human health.
Our analysis has a number of limitations. The analysis included rigid criteria for variant qualification and grouped variants at the most basic level, the gene. Future studies in this dataset can utilize more complex weighting algorithms as opposed to rigid cutoffs and can explore different ways of grouping rare variants, such as by gene family or by exon. Our study used a simple dominant model of inheritance, while recessive models and models that include gene–gene or gene–variant interactions will doubtless provide novel insights as well. Our study was also restricted to the CDS portions of the genome, and future work must expand further, especially to comprehensively analyze rare variants in non-coding regions. Although our initial explorations did not find utility in variance-component score and weighted analysis methods or methods that utilize variants beyond the coding regions (see Supplementary Note on our analyses utilizing the SKAT test and CADD scores), additional work focusing on these areas will likely identify novel associations, as they have proved useful in some prior studies. Finally, many phenotypes included in this study had a small number of cases, which reduced the power for discovery, and will no doubt become better powered as more of these large-scale population studies are completed.
This analysis presents one of the first forays into a new standard for human genetics research. As the sample sizes of cohorts with extensive phenotypic data and next generation sequencing grows, both through publicly available cohorts such as the UKB and population-based screening efforts such as the Healthy Nevada Project, we are now able to investigate the biological impact of rare variants with the same fine-tuned precision with which we currently assess the effects of common variants. A wealth of discoveries await us as we embark on this next phase of incorporating rare genome sequencing information into truly personalized medicine. We provide an interactive browser of our results as a resource to the human genetics community (https://ukb.research.helix.com/) to facilitate these discoveries.