Alan P. Tenney, Silvio Alessandro Di Gioia, Bryn D. Webb, Wai-Man Chan, Elke de Boer, Sarah J. Garnai, Brenda J. Barry, Tammy Ray, Michael Kosicki, Caroline D. Robson, Zhongyang Zhang, Thomas E. Collins, Alon Gelber, Brandon M. Pratt, Yuko Fujiwara, Arushi Varshney, Monkol Lek, Peter E. Warburton, Carol Van Ryzin, Tanya J. Lehky, Christopher Zalewski, Kelly A. King, Carmen C. Brewer, Audrey Thurm, Joseph Snow, Flavia M. Facio, Narisu Narisu, Lori L. Bonnycastle, Amy Swift, Peter S. Chines, Jessica L. Bell, Suresh Mohan, Mary C. Whitman, Sandra E. Staffieri, James E. Elder, Joseph L. Demer, Alcy Torres, Elza Rachid, Christiane Al-Haddad, Rose-Mary Boustany, David A. Mackey, Angela F. Brady, María Fenollar-Cortés, Melanie Fradin, Tjitske Kleefstra, George W. Padberg, Salmo Raskin, Mario Teruo Sato, Stuart H. Orkin, Stephen C. J. Parker, Tessa A. Hadlock, Lisenka E. L. M. Vissers, Hans van Bokhoven, Ethylin Wang Jabs, Francis S. Collins, Len A. Pennacchio, Irini Manoli & Elizabeth C. Engle
7817 Accesses, 2 Citations, 51 Altmetric
Abstract
Hereditary congenital facial paresis type 1 (HCFP1) is an autosomal dominant disorder of absent or limited facial movement that maps to chromosome 3q21-q22 and is hypothesized to result from facial branchial motor neuron (FBMN) maldevelopment. In the present study, we report that HCFP1 results from heterozygous duplications within a neuron-specific GATA2 regulatory region that includes two enhancers and one silencer, and from noncoding single-nucleotide variants (SNVs) within the silencer. Some SNVs impair binding of NR2F1 to the silencer in vitro and in vivo and attenuate in vivo enhancer reporter expression in FBMNs. Gata2 and its effector Gata3 are essential for inner-ear efferent neuron (IEE) but not FBMN development. A humanized HCFP1 mouse model extends Gata2 expression, favors the formation of IEEs over FBMNs and is rescued by conditional loss of Gata3. These findings highlight the importance of temporal gene regulation in development and of noncoding variation in rare mendelian disease.
Main
The noncoding human genome contains cis-regulatory elements (cREs) that can be bound by transcription factors (TFs) and act as cell-type-specific enhancers or silencers to define complex gene regulatory programs. Recent advances have revealed that cRE variants may cause rare disease; however, determination of the precise mechanism is difficult due to the need to study cREs in their relevant cellular and temporal context. Such studies are particularly challenging for developmental disorders where the fate of a small number of progenitors is defined by dynamic transcriptional states.
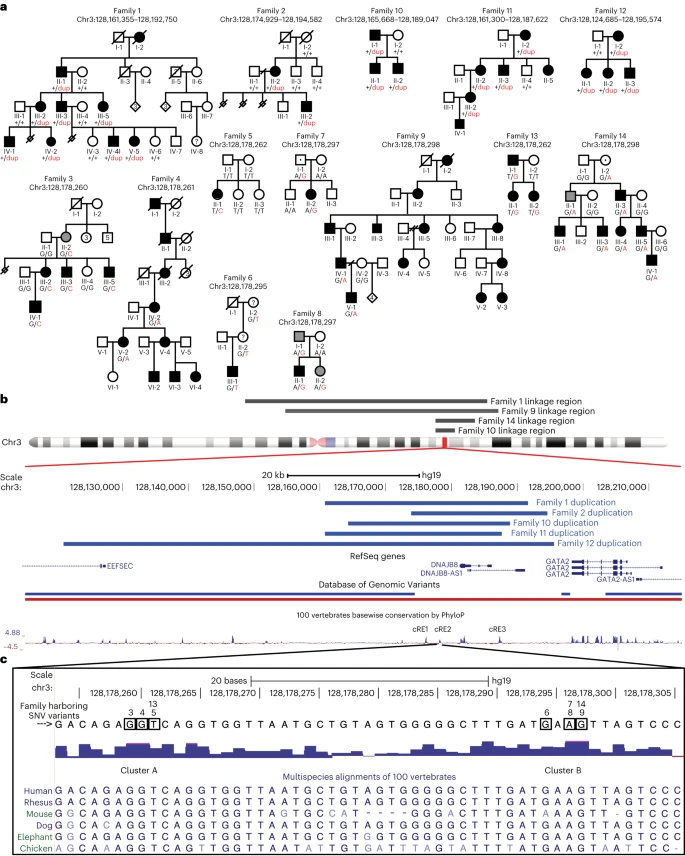
HCFP1 is a rare autosomal dominant disorder of absent or limited facial movement that was mapped to a 3-cM region of chromosome 3q21.2–22 (refs.). Neuropathology revealed a decreased number of FBMNs and facial nerve hypoplasia. Sequencing of genes in the critical region, including GATA2, did not identify pathogenic coding variants17.
In the present study, we report that HCFP1 results from noncoding variants within a cell-type-specific GATA2 regulatory region. We identified two adjacent clusters of noncoding SNVs that alter a conserved cRE (cRE2) and overlapping tandem duplications of cRE2 and the adjacent GATA2 enhancers, cRE1 and cRE3. We demonstrate that one cRE2 SNV cluster impairs binding of nuclear receptor subfamily 2 group F member 1 (NR2F1; COUP-TF1) and attenuates its repressive activity in a cell-specific manner. We show that GATA2, and its downstream effector GATA3 (refs.), are necessary to differentiate rhombomere 4 motor neurons (r4MNs) to IEEs but are dispensable for FBMN development. By contrast, a humanized cRE1 duplication mouse has ectopic expression of Gata2 in developing FBMNs and this phenotype is rescued by genetically ablating Gata3. This mechanism highlights the importance of tight temporal control of TF expression in a cell-type-specific manner during development and supports whole-genome sequencing (WGS) to identify noncoding variation underlying rare Mendelian disorders.
Results
We enrolled families and simplex cases with nonsyndromic congenital facial paresis (CFP, cohort 1 US-based study) and performed genome-wide single-nucleotide polymorphism (SNP) analysis and whole-exome sequencing (WES) in two large dominant pedigrees, family 1 (Fam1) and family 9. SNP-based multipoint parametric linkage analysis assumed autosomal dominant inheritance and full penetrance yielded maximum lod (logarithm of odds) scores suggestive of linkage at an overlapping 63-Mb chr3 region encompassing the previously reported HCFP1 locus (Fig. and Extended Data Fig.). WES analysis did not identify pathogenic coding variants within the suggestive regions of linkage in either family. To identify HCPF1 variants, we performed WGS from members of Fam1, Fam9 and seven additional HCFP pedigrees in cohort 1 (two vertical, one horizontal transmission and four simplex cases). Structural variation analysis revealed 31-kb and 20-kb overlapping tandem duplications within the HCPF1 locus in Fam1 and Fam2 (de novo), respectively (and Extended Data Fig.). We next analyzed WGS for SNVs or indels (insertions and deletions) within the Fam1/Fam2 ~18-kb minimum duplication region. Fam3, Fam7 and Fam9 each harbored a unique SNV within an ~270-bp, noncoding, conserved element (chr3:128,178,158–128,178,397; GRCh37/hg19). We resequenced and conducted double droplet PCR (ddPCR) of this element in the remaining cohort 1 probands: 2 pedigrees with vertical transmission, 4 sibling pairs and 31 simplex cases. SNVs were identified in dominant Fam4 and Fam8 and simplex Fam5 (de novo) and Fam6 (and Extended Data Fig.).
Discussion
We report that heterozygous noncoding SNVs and CNVs at the HCFP1 locus alter regulation of GATA2 and account for >90% of autosomal dominant, nonsyndromic CFP. Remarkably, the SNVs alter six nucleotides located in two clusters within a conserved noncoding region that we refer to as cRE2, located 3′ of DNAJB8 and GATA2. DNAJB8 is not a triplosensitive gene (pTriplo score 0.22) nor is it expressed in r4MNs or surrounding tissue in WT or cRE1dup/+ mice, excluding its involvement in HCFP1. Instead, our data support cRE2 as a tissue-specific regulatory element to which NR2F1 binds, restricting r4MN GATA2 expression to developing IEEs.