Natsuhiko Kumasaka, Raghd Rostom, Ni Huang, Krzysztof Polanski, Kerstin B. Meyer, Sharad Patel, Rachel Boyd, Celine Gomez, Sam N. Barnett, Nikolaos I. Panousis, Jeremy Schwartzentruber, Maya Ghoussaini, Paul A. Lyons, Fernando J. Calero-Nieto, Berthold Göttgens, Josephine L. Barnes, Kaylee B. Worlock, Masahiro Yoshida, Marko Z. Nikolić, Emily Stephenson, Gary Reynolds, Muzlifah Haniffa, John C. Marioni, Oliver Stegle, Tzachi Hagai & Sarah A. Teichmann
9205 Accesses, 136 Altmetric
Abstract
Common genetic variants across individuals modulate the cellular response to pathogens and are implicated in diverse immune pathologies, yet how they dynamically alter the response upon infection is not well understood. Here, we triggered antiviral responses in human fibroblasts from 68 healthy donors, and profiled tens of thousands of cells using single-cell RNA-sequencing. We developed GASPACHO (GAuSsian Processes for Association mapping leveraging Cell HeterOgeneity), a statistical approach designed to identify nonlinear dynamic genetic effects across transcriptional trajectories of cells. This approach identified 1,275 expression quantitative trait loci (local false discovery rate 10%) that manifested during the responses, many of which were colocalized with susceptibility loci identified by genome-wide association studies of infectious and autoimmune diseases, including the OAS1 splicing quantitative trait locus in a COVID-19 susceptibility locus. In summary, our analytical approach provides a unique framework for delineation of the genetic variants that shape a wide spectrum of transcriptional responses at single-cell resolution.
Main
The innate immune response is a cell-autonomous program that induces an antiviral state in infected and nearby cells and alerts the immune system of the invading pathogen1. Dysregulation of this response can affect a wide range of inflammatory and autoimmune diseases and determine the outcome of infection2,3,4,5,6. Common genetic variants have been shown to modulate transcriptional responses to various viral and bacterial stimuli, and to contribute to disease onset and progression7,8,9,10,11. Most past gene-expression-focused studies of this program are based on bulk RNA-sequencing (RNA-seq) technologies, which do not fully elucidate the continuous dynamics of transcriptional changes during the innate immune response. Single-cell genomic technologies are powerful approaches to study cell heterogeneity and transcriptional variability across cells12. Furthermore, by utilizing single-cell RNA-seq (scRNA-seq) profiling of tissues composed of several cell lineages, previous studies have successfully performed genetic association mapping of cell-type-specific expression13,14,15,16,17,18,19.
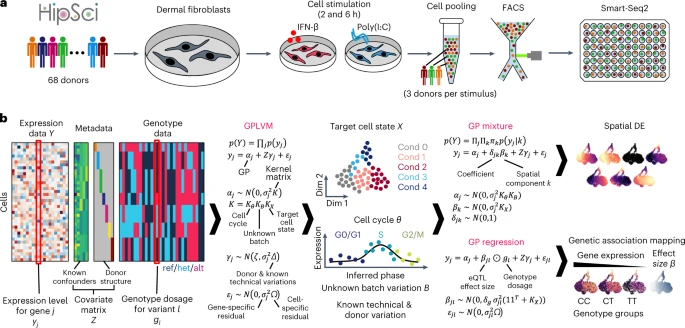
We here use full-length scRNA-seq of dermal fibroblasts from different human individuals, challenged with immune stimuli. Based on the pseudo-temporal reconstruction of these data, we map the transcriptional variation of the innate immune response at single-cell resolution. This provides the foundation for superimposing human genetic variation onto the transcriptional dynamics of this response. To this end, we develop a statistical approach based on a Gaussian process (GP) latent variable model (GPLVM)20,21 called GASPACHO (GAuSsian Processes for Association mapping leveraging Cell HeterOgeneity). This allows us to identify expression quantitative trait loci (eQTLs) that manifest at different stages of the response to stimuli.
We find more than a thousand eQTLs, hundreds of which are colocalized with known risk loci of diverse autoimmune and infectious diseases. We perform fine-mapping of the OAS1 locus, associated with COVID-19, to reveal the imbalanced expression of OAS1 and OAS3 genes during the antiviral innate immune response. We further integrate these data with eQTLs from a COVID-19 patient cohort dataset of peripheral blood mononuclear cell (PBMC) scRNA-seq22, as well as with scRNA-seq data of infected nasal epithelial cells from 33 patients with COVID-19 (ref. 23).
Overall, our study illustrates how coupling single-cell transcriptomics with a cutting-edge statistical approach can identify dynamic effects of human trait-associated genetic variants in different contexts of activation of antiviral innate immunity and, in general, in diverse cellular dynamic processes.
Results
Cell stimulation to study antiviral responses in fibroblasts
To study the innate immune expression program that is triggered upon viral infection, we exposed primary dermal fibroblasts from 68 donors from the Human Induced Pluripotent Stem Cell Initiative (HipSci)24 to two stimulants: (1) Poly(I:C), a synthetic double-stranded RNA (dsRNA) that is rapidly recognized by viral sensors and elicits primary antiviral and inflammatory responses; and (2) interferon-β (IFN-β), a cytokine that upregulates a secondary wave of response in both infected and bystander cells, and shifts the cells into an antiviral mode, where hundreds of interferon-stimulated genes (ISGs) are upregulated to contain the infection.
We collected cells exposed to each of the two stimuli after 2 and 6 h of stimulation (Fig. 1a). Following this, scRNA-seq profiling was performed using a plate-based full-length transcript approach (Methods). After quality control, 22,188 high-quality cells were obtained across 128 plates, with each plate containing cells from three donors (Fig. 1a). The donor identity for each cell was inferred from scRNA-seq read data using known genotypes made available by HipSci (Extended Data Fig. 1a and Methods). Preliminary analysis showed that our data display high cell-to-cell variability in gene expression both within and across donors, as observed in previous studies by us and others25,26,27. In fact, our data were confounded by various technical and biological factors, including library preparation in different batches, and cell cycle effects (Extended Data Fig. 1b). The complex nature of these data, along with their confounders, motivated us to develop an approach that reveals the genetic and physiologically relevant variation, while computationally masking confounding factors.
Discussion
In this work we developed GASPACHO, a statistical framework that fulfils two tasks: firstly, to infer the trajectory of gene expression over a dynamic process, and secondly, to model nonlinear dynamic genetic effects in every individual cell. Using GASPACHO, we integrated scRNA-seq data from fibroblasts from 68 donors triggered by innate immune stimuli and obtained a low-dimensional gene expression space representing the response dynamics across stimulated cells. This approach provides us with a unique map of interindividual transcriptional variation at single-cell resolution, which was often linked with noncoding regulatory regions (such as TF binding sites), previously profiled during fibroblast stimulation27. This approach discovered 1,275 eQTL loci, of which 988 were colocalized with one or more GWAS loci of autoimmune and infectious diseases, including COVID-19 at the OAS1 locus. We also found 1,607 colocalizations shared with the Open Targets colocalizations database, of which 615 also overlap with the GTEx fibroblast colocalization analysis. We note here that some of the colocalizations in the Open Targets database are likely to be missed because Open Targets has only tested colocalizations against genome-wide significant loci in GWAS traits (P < 5 × 10–8), while we tested colocalizations for all GWAS loci (regardless of their P values) overlapping with 1-megabase (Mb) cis-windows of genes tested for eQTL mapping.
Previous studies used scRNA-seq profiling of tissues composed of several cell types from dozens to hundreds of donors, and performed genetic association mapping of cell-type-specific expression by using a pseudo-bulk approach13,14,15,16,17,18,19. GASPACHO, as well as cellRegMap33, allows mapping of dynamic genetic effects of gene expression in individual cells. These two approaches are particularly suitable when considering a continuous genetic effect along cellular states rather than several discrete cell populations or states. GASPACHO and CellRegMap both incorporate context-specific donor (donor by context interaction) effects to adjust for dynamic genetic effects, for a better statistical calibration (because dynamic cellular states, such as immune responses, often vary between donors due to environmental and trans genetic effects). However, CellRegMap and GASPACHO differ in their model assumptions on genetic effects: CellRegMap assumes linearity on context-specific genetic (genotype by context interaction) effects, while GASPACHO assumes that those effects are nonlinear, suggesting GASPACHO is more flexible, yet computationally intensive (section 3 of the Supplementary Notes). Therefore, further studies are required to implement faster GP regression in modern computational environments, such as GPU. Lastly, the GPLVM implemented in GASPACHO is currently applicable for rapid analysis of dozens of thousands of cells. An accelerated version of GPLVM will be needed in the future for scaling, and a cutting-edge Bayesian inference technique, such as the stochastic variational inference implemented in GPy, should be able to achieve this goal.
The innate immune response is a genetic program that is elicited by most cells invaded by pathogens; however, the response varies between infected cells in terms of magnitude, the specific set of regulated genes and their cellular fate. This variability is observed both between cells originating from different lineages and between individual cells from the same homogenous cellular population25,26,27. Furthermore, genetic variation has been shown to significantly modulate the innate immune response in many previous studies and its dynamics are thought to be nonlinear4,6,7,8,9,35. GASPACHO is thus particularly useful to study how genetic effects are associated with different stages and cellular trajectories during this response, as demonstrated, for example, in our analysis of primary and secondary response genes. Furthermore, we also observe eQTLs that appear only during stimulation, as previously suggested using bulk RNA-seq4,6,7,8,9,35, but without the need to partition samples into discretized conditions.
Our in vitro immune stimulation of dermal fibroblasts can be used as a model system to study genetic effects in innate immune responses in primary cells. Using this analysis, we detected 6,327 colocalizations between eQTLs and various autoimmune and infectious disease GWAS loci. While fibroblasts are not the primary cellular target of SARS-CoV-2 infection (which mostly targets epithelial cells), we detected a colocalization between OAS1 eQTL and COVID-19 GWAS locus, which we then also found in PBMCs from patients with COVID-19. This colocalization was also previously found using bulk RNA-seq41. Our findings suggest an association between a particular risk variant (rs10774671) and COVID-19 infection and severity, and that this risk allele may generate alternative isoforms of the OAS1 gene in nonclassical monocytes in peripheral blood. We further found that these alternative isoforms are expressed in nasal epithelial cells from a set of patients with COVID-19 carrying the alternative allele. Since the alternative allele is also a risk allele in COVID-19 GWAS, this implies that these OAS1 RNA splicing isoforms may be associated with impaired OAS1 protein expression and viral clearance in host cells, as previously suggested in other viral diseases40,42. Interestingly, we also observe a colocalization in this locus between OAS3 eQTL and COVID-19 GWAS locus; however, in this case, the alternative allele of rs10774671 is linked to an increase in the OAS3 gene expression level. Further studies are needed to mechanistically determine the impact of OAS3 expression on SARS-CoV-2 infection.
In summary, our study demonstrates how coupling single-cell transcriptomics with a statistical approach can identify dynamic nonlinear effects of genetic variants across cellular contexts.